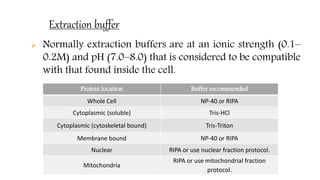
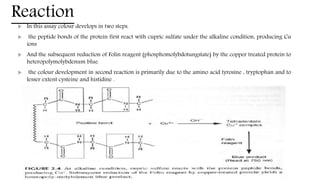
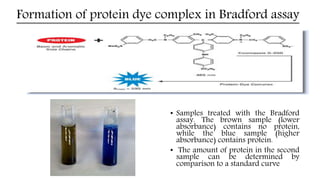
The document discusses methods for preparing tissue or cell extracts for protein separation and analysis. It describes various cell lysis buffers and their uses depending on the protein location. It also discusses steps to inhibit protein degradation during extraction, such as using protease inhibitors and reducing agents. The document compares the Lowry and Bradford methods for estimating protein concentration, noting the principles, advantages, and disadvantages of each. It also discusses the importance of trichloroacetic acid precipitation to separate proteins from interfering substances.













![Advantages and disadvantages
Advantages
Fast and inexpensive
Highly specific for protein
Very sensitive [1-20 µg (micro assay) 20-200 µg (macro assay)]
Compatible with a wide range of substances
Extinction co-efficient for the dye-protein complex is stable over 10 orders of
magnitude (assessed in albumin)
Dye reagent complex is stable for approximately one hour
Disadvantages
Absorbance spectra of the two coomassie brilliant blue G-250 species partially
overlap making the standard curve.
Non-linear standard curve over wide ranges
Response to different proteins can vary widely, choice of standard is very
important
It is also inhibited by the presence of detergents.
The Bradford assay is linear over a short range, typically from 0 µg/ml to
2000 µg/ml, often making dilutions of a sample necessary before analysis.](https://image.slidesharecdn.com/cellbiologypractical-171105071458/85/PREPERATION-F-CELL-EXTRACT-18-320.jpg)










































































