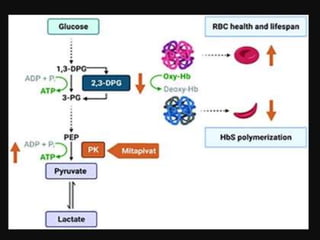
Mitapivat is an oral activator of pyruvate kinase that has shown promise in treating pyruvate kinase deficiency and other hemolytic anemias. Clinical trials have found it to be well-tolerated and able to significantly increase hemoglobin levels in patients with pyruvate kinase deficiency or thalassemia. Ongoing phase 2 and 3 trials are investigating its efficacy and safety for reducing transfusions in thalassemia and decreasing symptoms in sickle cell disease.