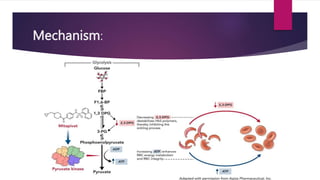
Thalassemia is a group of hemoglobinopathies leading to chronic anemia due to ineffective erythropoiesis, predominantly affecting ATP levels in red blood cells. Mitapivat, an allosteric activator of pyruvate kinase, improves ATP production in RBCs, which is crucial for managing anemia. Common genetic mutations, particularly deletions in the α-globin gene, play a significant role in the severity of α-thalassemia.













![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)











































