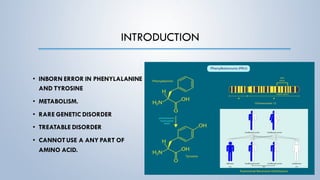
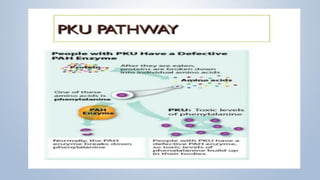
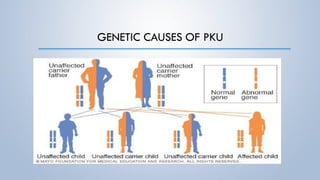
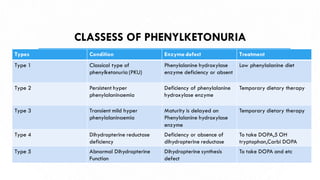
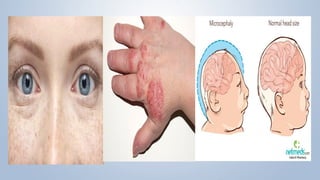
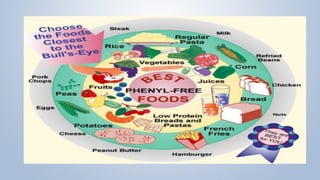
This document discusses phenylketonuria (PKU), a rare genetic disorder caused by mutations in the PAH gene that prevents the conversion of phenylalanine to tyrosine. This leads to a toxic buildup of phenylalanine in the body. PKU is diagnosed through newborn screening and confirmed with blood tests measuring phenylalanine levels. Treatment involves a lifelong low-phenylalanine diet to prevent intellectual disabilities and other health issues. If left untreated, PKU can cause issues such as low IQ, seizures, behavioral problems, and eczema. Early diagnosis and dietary treatment can help patients with PKU live healthy lives.