WHAT IS REGULATORYAFFAIRS?
◦ A regulatory affairs is a profession which act as interface
between pharmaceutical industry and drug regulatory authority
around the world.
COMPANY
PRODUCT
REGULATORY
AFFAIRS
REGULATORY
AUTHORITY
3.
GOAL’S OF RA
◦Protection of Human health
◦ Ensuring safety ,efficacy and quality of drugs
◦ Ensuring accuracy of product information
4.
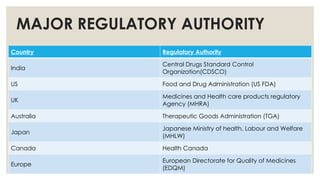
MAJOR REGULATORY AUTHORITY
CountryRegulatory Authority
India
Central Drugs Standard Control
Organization(CDSCO)
US Food and Drug Administration (US FDA)
UK
Medicines and Health care products regulatory
Agency (MHRA)
Australia Therapeutic Goods Administration (TGA)
Japan
Japanese Ministry of health, Labour and Welfare
(MHLW)
Canada Health Canada
Europe
European Directorate for Quality of Medicines
(EDQM)
5.
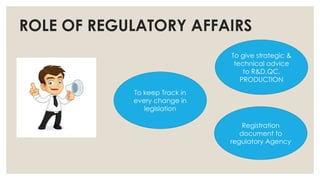
ROLE OF REGULATORYAFFAIRS
Registration
document to
regulatory Agency
To keep Track in
every change in
legislation
To give strategic &
technical advice
to R&D,QC,
PRODUCTION
MASTER FORMULA RECORD
◦Master Formula Record (MFR) is a master document for any
pharmaceutical product.
◦ MFR contains all information about the manufacturing process
for the product.
◦ MFR is prepared by the research and development team of the
company.
◦ MFR is used as reference standard for preparing batch
manufacturing record (BMR) by manufacturing units.
◦ MFR is also called Master Manufacturing Record, Master
Production Record.
8.
◦ “A documentor set of documents specifying the starting material with their quantities
and the packaging materials, together with a description of the procedure and
precautions required to produce a specified quantity of a finished product as well as the
processing instructions, including the in-process controls”.
◦ Master Formula Record (MFR) is a master document for any pharmaceutical product.
◦ There shall be Master Formula Records relating to all manufacturing procedures for each
product and batch size to be manufactured.
◦ These shall be prepared and endorsed by the competent technical staff i.e., head of
production and quality control.
◦ A Master Formula Record is either prepared based upon experience of impotent qualified
staff like manufacturing chemist or analytical chemist or prepared based upon batch
manufacturing record of a batch size.
9.
MASTER FORMULA RECORDCONTAIN’S
Product Details
◦ Name, logo and address of
the manufacturing company
◦ Dosage form name (brand
and generic name)
◦ Product code and Label
claim of all ingredients
◦ Product description like
Batch size, Pack size and
packing style
◦ Shelf life
◦ Storage conditions
◦ MFR number and date
◦ Authorization by the production
and quality assurance head
Need of Documentation:
1.Defines specifications and procedures for all materials and methods of manufacture and control
2. Control of Process - Ensures all staff knows what to do and when to do it.
3. Ensure that authorized persons have all information necessary for release of product
4. Ensures documented evidence, traceability, provide records and audit trail for investigation
5. Ensures availability of data for validation, review and statistical analysis.
6. To improve performance
7. Regulatory requirements
15.
Importance of Documentation
1.It provides necessary working details
2. Reduces the risk of mistake
3. Help in tracing the deviation from the expected yield
4. They help in decreasing the batch-to-batch variation so that quality of product is kept
within the limits of acceptability
5. Considered as the history of batch operations
6. Self-inspection of procedure
DRUG MASTER FILE
◦It is a submission to USFDA or to concerned regulatory authority,
that may be used to provide confidential and detailed
information about manufacturing, processing ,packaging or
storing of one or more human drugs.
◦ DMF is not mandatory by law or FDA regulation.
◦ DMF is submitted by API manufacturers.
◦ The information in DMF is used to support NDA,ANDA,IND.
◦ It is a submission that indicates that product of the company is a
quality product and meets the required standards.
18.
TYPES OF DRUGMASTER FILES
In case of drug substance (DS), summarize all significant steps in the manufacturing and controls of the
drug intermediate or substance. Whereas, in case of drug product (DP), manufacturing procedures and
controls for finished dosage forms should ordinarily be submitted in an IND, NDA, ANDA, or Export
Application.
19.
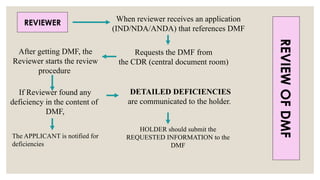
REVIEWER When reviewerreceives an application
(IND/NDA/ANDA) that references DMF
Requests the DMF from
the CDR (central document room)
After getting DMF, the
Reviewer starts the review
procedure
If Reviewer found any
deficiency in the content of
DMF,
The APPLICANT is notified for
deficiencies
DETAILED DEFICIENCIES
are communicated to the holder.
HOLDER should submit the
REQUESTED INFORMATION to the
DMF
REVIEW
OF
DMF
DISTRIBUTION RECORDS
◦ Distribution:The division and the movement of pharmaceuticals
products from the premises of the manufacturer to the end user
or to an intermediate point by means of various transport
methods.
◦ Distribution records: Are written data related to distribution of
drug products from manufacturer to the distributor.
22.
DISTRIBUTION PROCEDURE
◦ Aprocedure whereby the oldest approved stock of a drug
product is distributed first.
◦ It must be constructed and procedures established to facilitate
recall of defective product.
◦ The manufacturer must maintain records of all distribution
transactions involving in process or finished goods.
◦ Computerized tracking systems are most common.
23.
DISTRIBUTION RECORDS SHOULD
CONTAIN
◦Name
◦ Strength of the product
◦ Name and address of consignee
◦ Date and quantity shipped
◦ Control number of drug product
GENERIC DRUG
◦ Ageneric drug is “ a drug product that is comparable to/
bioequivalent to brand/innovator drug in dosage form, strength,
route of administration, quality & performance characteristics”.
◦ After the expiry of the patent or marketing rights of the patent
drug , generic drugs are marketed.
26.
PRODUCT DEVELOPMENT
◦ PRODUCT: A product is something sold by an enterprise to its
customers.
◦ PRODUCT DEVELOPMENT : Product development is the set of
activities beginning with the perception of a market opportunity
and ending in the production , sale and delivery of a product.
27.
GENERIC PRODUCT DEVELOPMENTPROCESS
◦ The input of the process is a mission statement and the output of
the process is the product launch.
◦ MISSION STATEMENT :
Identifies the target market for the product , provides a basic
functional description of the product , and specifies the
business goals of the effort ; results from well executed product
planning phase.
◦ PRODUCT LAUNCH :
Occurs when the product becomes available for purchase in
the market place.
28.
HATCH WAXMAN ACT
◦It is otherwise called as “Drug Price competition & Patent term
Restoration Act” .
◦ Established in 1984.
◦ The main objective is
- to reduce the cost
- to make available more low cost generic drugs
- Motivating the generic drug Manufacturer
◦ Generic drug manufacturers files ANDA that incorporates safety
and effectiveness data submitted by original pioneer drug
manufacturer and adds only bioequivalence study.
30.
PROVISIONS OF THEACT
◦ Paragraph I: that such patent information has not been filed
◦ Paragraph II: that such patent has expired
◦ Paragraph III: of the date on which such patent will expire
◦ Paragraph IV: that such patent is invalid or will not be infringed by
the manufacture, use, or sale of the new drug for which the
application is submitted
31.
ORANGE BOOK
◦ Containsthe list of all FDA approved Drug products
◦ It is updated monthly.
CODE OF FEDERALREGULATIONS (CFR)
◦ CFR is the codification of the general & permanent rules and regulations
(also called as Administrative law) published in the federal register by the
executive departments & agencies of the federal government of the united
states.
◦ It is divided into 50 titles that represent broad areas.
◦ Each title is further divided into chapters, subchapters, parts, and sections.
◦ Example: 21 CFR 310.502 Revised as of April 1, 1997
◦ Title: 21
◦ Part: 310
◦ Section: 502
◦ Year: 1997
◦ There are a number of electronic sources for accessing CFR.
34.
NOTABLE SECTIONS
◦ 11Electronic records and electronic signature related.
◦ 50 Protection of human subjects in clinical trials.
◦ 54 Financial Disclosure by Clinical Investigators.
◦ 56 Institutional Review Boards that oversee clinical trials.
◦ 58 Good Laboratory Practices (GLP) for nonclinical studies.
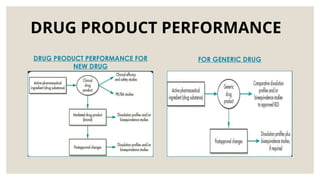
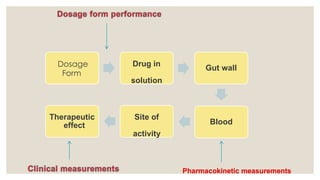
DRUG PRODUCT PERFORMANCE
(DPP)
◦DPP is defined as the release of the drug substance leading to
bioavailability of the drug substance.
◦ Assessment of DPP is important since bioavailability is related both
to the pharmacodynamics response and to adverse effects.
◦ DPP – determined by in-vivo bioequivalence studies or in-vitro by
comparative drug dissolution studies.
37.
DRUG PRODUCT PERFORMANCEIN-VITRO
* Dissolution and drug release tests are conducted in-vitro to measure how quickly and how much of a
drug substance dissolves or released from a drug product, typically in an aqueous medium under
specific conditions.
* These test provide critical data throughout the drug development process.
• The dissolution test serve as a key quality control procedure, ensuring batch consistency and
identifying any variability in composition and manufacturing process.
• When linked to in-vitro performance, the dissolution and drug release to provide how well the drug
perform in the body.
• Ideally, the in-vitro dissolution method should closely simulate the drug release profile in -vivo and
should distinguish between formulation with varying release characteristics.
• The primary goal is to develop a dissolution test that can differentiate between acceptable and
suboptimal formulation by observing differences in dissolution rates under identical test conditions.
38.
◦ A well-designeddissolution test should be sensitive enough to detect changes in formulation,
manufacturing process, or the physical and chemical properties of the drug, such as particle size,
polymorphism and surface area.
◦ Dissolution testing is often required for routine batch testing and assessing changes made during scale
up or after regulatory approval (SUPAC) for marketed products.
◦ For minor changes, the effect on in-vivo performance can be evaluated by comparing dissolution
profiles of the product before and after the modification using the approved dissolution method or
testing under different pH condition.
◦ Major post approval changes in the manufacturing process may require bioequivalence studies,
although this requirement may be waived if there is an acceptable in vitro- in vivo correlation.
◦ The dissolution test should be robust, reproducible and transferable between laboratories to ensure
consistency.
39.
DRUG PRODUCT PERFORMANCEIN-VIVO:
* In-vivo drug product performance refers to the release of the drug substance
from the product and its subsequent bioavailability.
* Evaluating in-vivo performance is critical because bioavailability affects the
pharmacodynamic response and any potential adverse events.
* Therefore, performance tests link the quality of a drug product to its clinical
safety and efficacy.
* Bioavailability and bioequivalence studies are key measures of a drug’s in-
vivo
performance.
• These studies are essential for both new and generic drug development.
40.
Bioavailability studies assessthe impact of changes in the drug’s physicochemical
properties, formulation and manufacturing process on performance. They ensure the new
drug product performs similarly to the original formulation used in clinical safety and
efficacy trails.
Bioequivalence studies compare the bioavailability of test drug product to that of a
reference product with the same active ingredients.
In general, in-vivo bioequivalence is demonstrated in healthy volunteers, though under
specific circumstances, in-vitro methods like comparative dissolution profiles can be used.
During drug development, bioequivalence studies compare formulation across early and
late clinical trails, between formulations and those intended for market or among different
strengths of a product.
Bioequivalence studies also support new formulations of previously approved drugs, such
as fixed dose combinations or modified release drugs.
41.
Post approval in-vivobioequivalence studies may be necessary for major formulation,
manufacturing , or site changes, to ensure the marketed drug product continues to perform as
expected.
The drug product approved by the FDA may differ from the original formulation tested in
clinical trials and any post approval changes known as SUPAC require the manufacturer to
demonstrate equivalent drug performance.
For generic drugs, bioequivalence studies are essential for regulatory approval, as clinical
safety and efficacy trials are not typically conducted for generics.
Generic drug manufacturers must demonstrate that their products are pharmaceutically
equivalent, bioequivalent and therapeutically equivalent to the reference brand name drug ,
typically through in-vivo bioequivalence studies under both fasting and fed conditions.
METHOD FOR ASSESSING
BIOAVAILABILITY& BIOEQUIVALENCE
In-vivo Measurement
of active moiety or
moieties in biological
fluids
• Plasma drug
concentration
• Time for Peak
Plasma
concentration
(Tmax)
• Peak plasma
concentration
(Cmax)
• Area under the
plasma drug
concentration-time
curve(AUC)
Urinary Drug
Excretion
• Cumulative
amount of drug
excreted in the
urine(Du)
• Rate of drug
excretion in the
urine(dDu/dt)
• Time for maximum
urinary excretion (t)
In-vivo
Pharmacodynamic
comparison
• Maximum
pharmacodynamic
effect (Cmax)
• Time for Maximum
pharmacodynamic
effect
• Area under the
curve (AUC)
• Onset time for
Pharmacodynamic
effect
In-vivo studies
• Comparative drug
dissolution
Clinical Endpoint
study
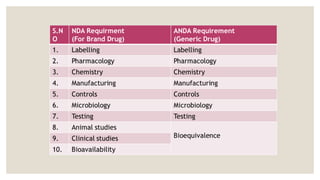
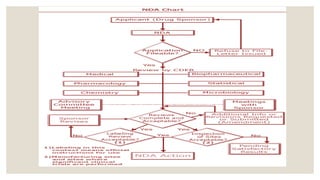
NDA (NEW DRUGAPPLICATION)
• The vehicle through which drug sponsors formally propose that
the regulatory body approve a new pharmaceutical for sale
and marketing.
• The data gathered during the animal studies and human
clinical trials of an Investigational new product become part of
the NDA.
47.
FUNDAMENTALS OF NDA
SUBMISSION
◦Index
◦ Summary
◦ Chemistry, Manufacturing, and
Control
◦ Samples, Method Validation
Package, and Labeling
◦ Nonclinical Pharmacology and
Toxicology
◦ Human Pharmacokinetics and
Bioavailability
◦ Microbiology (for anti-microbial
drugs only)
◦ Clinical Data
◦ Safety Update Report (typically
submitted 120 days after the NDA's
submission)
◦ Statistical
◦ Case Report Tabulations
◦ Case Report Forms
◦ Patent Information
49
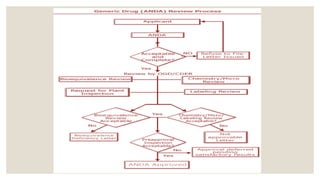
ANDA(ABBREVIATED NEW DRUG
APPLICATION)
◦ Abbreviated new drug application (ANDA) is an application for a U.S.
generic drug approval for an existing licensed medication or
approved drugs.
◦ A generic drug product is one that is comparable to an innovator
drug product in its dosage form , strength , quality etc.
51.
51
CONTENT AND FORMATOF AN ABBREVIATED
APPLICATION
1. Application form
2. Table of contents
3. Basis for abbreviated new drug application submission
4. Conditions of use
5. Active ingredients
6. Route of administration, dosage form and strength
7. Bioequivalence
8. Labeling
9. Chemistry, manufacturing and controls.
10. Samples
11. Patent certification.
BIOEQUIVALENCE AND DRUG
PRODUCTASSESSMENT
By World Health Organization (WHO):
Two pharmaceutical products are bioequivalent if they are
pharmaceutically equivalent , and their bioavailabilities, in terms
of rate (Cmax and tmax) and extent of absorption (area under
the curve), after administration of the same molar dose under the
same conditions, are similar to such a degree that their effects can
be expected to be essentially the same.
54.
NEED OF BIOEQUIVALENCE
◦The need of bioequivalence studies is increasing due to the
large growth of the production and consumption of generic
product
Bioequivalence studies are conducted if there is :
◦ A risk of bio inequivalence or
◦ A risk of pharmacotherapeutic failure
◦ No clinical studies have been performed in patient with the
generic product to support its efficacy and safety
55.
TYPES OF EQUIVALENCE
1.Chemical equivalence
Two or more drug product contain same labelled chemical in a same amount.
2. Pharmaceutical equivalence
Two or more drug are identical in strength, quality, purity, content uniformity,
disintegration and dissolution.
3. Therapeutic equivalence
Indicate that two or more drug product that contain the same therapeutically
active ingredient elicit identical pharmacological effect and control the disease
to the same extent.
4. Bioequivalence
It is a relative term which denotes that the drug substance in two or more
identical dosage form, reaches the systemic circulation at the same relative
rate and relative extent.
56.
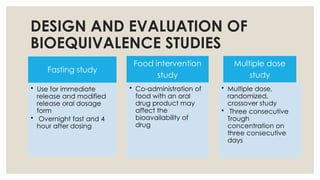
DESIGN AND EVALUATIONOF
BIOEQUIVALENCE STUDIES
Fasting study
• Use for immediate
release and modified
release oral dosage
form
• Overnight fast and 4
hour after dosing
Food intervention
study
• Co-administration of
food with an oral
drug product may
affect the
bioavailability of
drug
Multiple dose
study
• Multiple dose,
randomized,
crossover study
• Three consecutive
Trough
concentration on
three consecutive
days
57.
TYPE OF DESIGN
1.Completerandomized design
All treatment are randomly allocated among all experimental
subject.
example: if there is 20 subjects, number them from 1 to20. Random select
non repeating number
2. Randomized block designs
Subjects are sorted into homogenous group called blocks.
Method
Subjects having similar background characteristics are formed as blocks
58.
EVALUATION OF DATA
Analytical data
-Analytical method for measurement of drug must be validated for
accuracy, precision, sensitivity and specificity.
-More then one method during bioequivalence study may
not be valid because different methods may yield different
values.
BIOWAIVERS
o The termBiowaiver is applied to a drug regulatory approval process when a dossier
(application) is approved based on the evidence of Bioequivalence.
o The biowaiver means that the in vivo bioavailability and bioequivalence studies may
be waived (i.e not necessary for the product approval)
o In 1995 , US department of Health and Human Services , and US-FDA started the
Biopharmaceutical Classification System ,with the aim of granting so called
Biowaivers for SUPAC.
o Applicant can request biowaiver for immediate release product based on an
approach termed the biopharmaceutics classification system BCS (32).
61.
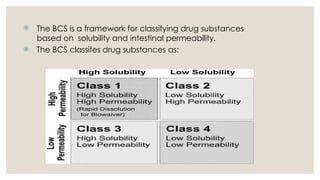
The BCSis a framework for classifying drug substances
based on solubility and intestinal permeability.
The BCS classifes drug substances as:
62.
SCALE-UP PROCESS APPROVALCHANGES
◦ The scale-up process approval changes made after approval in
the composition, Manufacturing process, mfg. equipment &
change of site have become known as Scale-up and Post
Approval Changes.
◦ Abbreviated as SUPAC.
NDA → Larger Batch Size
ANDA → Larger Batch Size
63.
SCALE-UP POST APPROVAL
CHANGES
◦The FDA has issued various guidance for SUPAC. They are
SUPAC – IR
(For Immediate-release solid oral dosage form)
SUPAC – MR
(For Modified-release Solid oral dosage form)
SUPAC – SS
(For Non-sterile semisolid dosage form such as creams, ointments,
gels & lotions)
64.
POST MARKETING SURVEILLANCE
(PMS)
◦PMS is the practice of monitoring the safety of a pharmaceutical drug
or medical device after it has been released on the market.
◦ It is an important part of the science of Pharmacovigilance.
Objective:
To develop information about drug effects under customary
condition of drug use.
To assess a known serious risk related to the use of drug
◦ Advantages:
Helps to detect rare ADR’s
Drug interaction
65.
METHODS OF SURVEILLANCE:
Fourtypes of studies are generally are used to identify drug effects
1. Controlled clinical trials
2. Spontaneous & Voluntary recording
3. Cohort studies
4. Case Control studies
66.
SOURCES OF PMS
1.Customer surveys
2. Literature reviews
3. Expert user groups
4. Customer complaints
5. The media
67.
OUTSOURCING BA ANDBE TO CRO
Definition
◦ Contract research organisation (CRO) is an entity that extends
support to pharmaceutical, biotechnology and medical devices
industry in the form of research services outsourced on a contract
basis.
◦ A CRO may offer such services like:
Biopharmaceutical
development
Clinical &
preclinical
research
Clinical trial
Biological assay
development
Pharmacovigilance
68.
OUTSOURCING BA ANDBE
TO CRO
◦ Outsourcing is the business practice & hiring a party outside a
company to perform services & create goods that traditionally
were performed in-house by the company’s own employee &
staff.
◦ It is generally done to
i. To reduce the costs and
ii. Improving the efficient resources within a company
Eg:
Outsourcing is Bioavailability, Bioequivalence, Research &
Development Department etc.