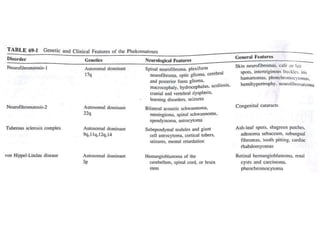
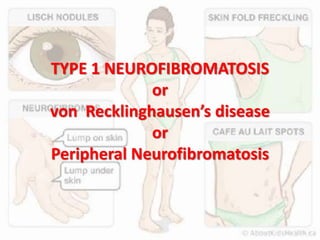
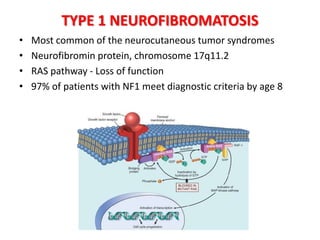
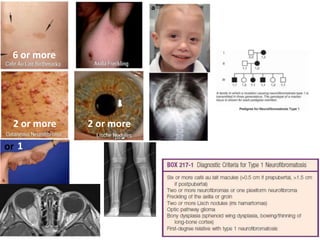
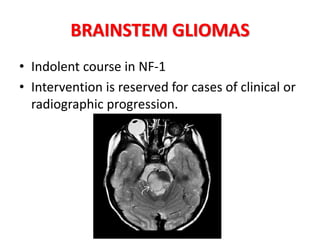
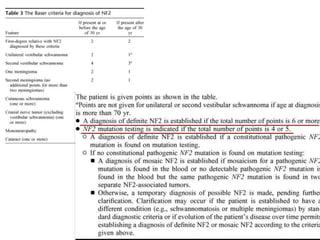
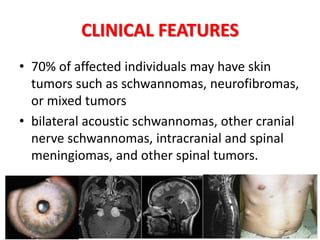
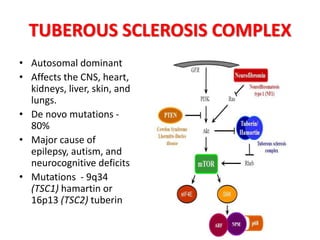
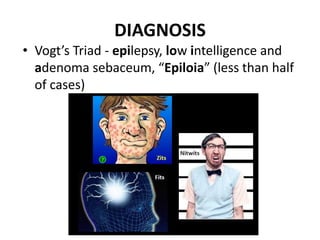
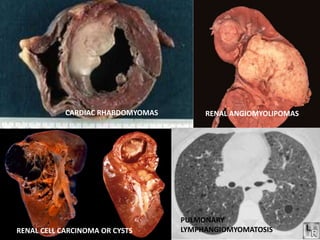
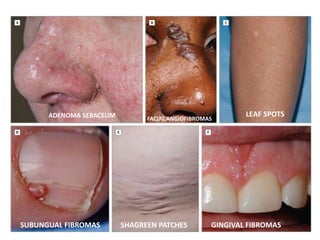
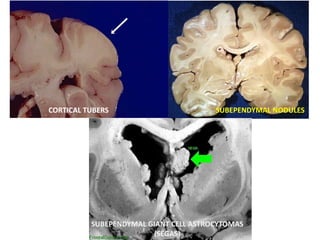
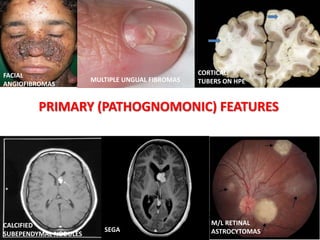
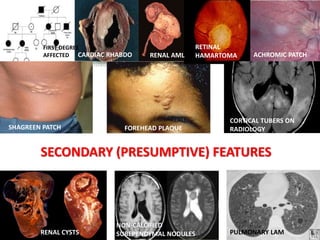
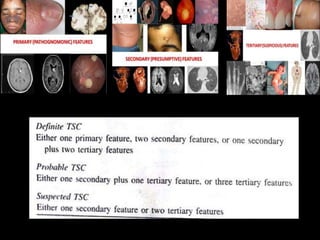
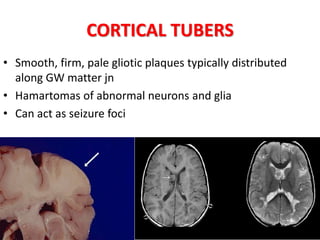
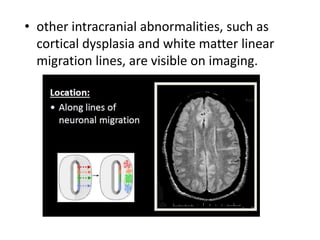
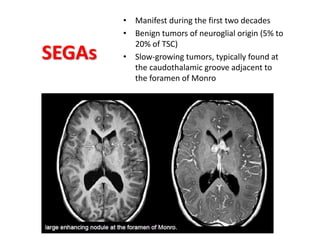
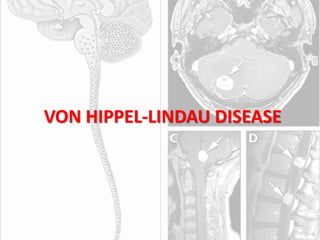
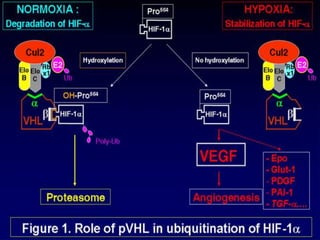
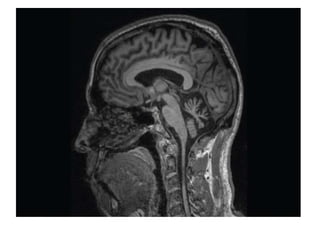
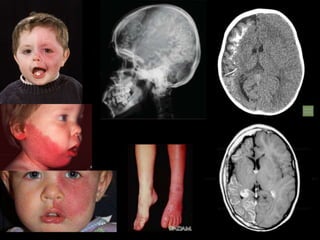
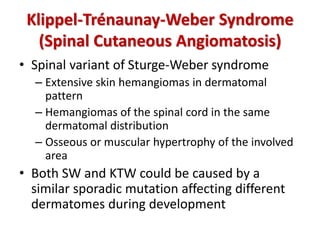
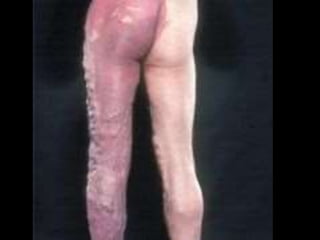
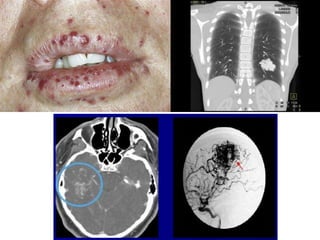
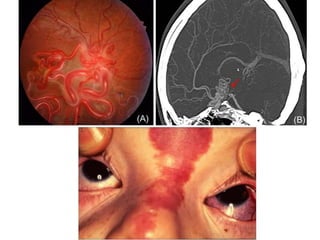
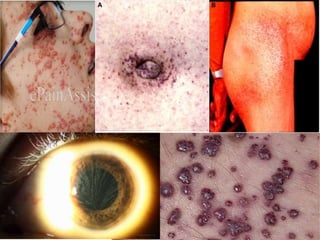
This document discusses several neurocutaneous disorders known as phakomatoses that are characterized by abnormalities affecting the skin, eyes, and nervous system. It provides detailed information on type 1 and type 2 neurofibromatosis, tuberous sclerosis complex, Von Hippel-Lindau disease, and several neurocutaneous angiomatoses including Ataxia-Telangiectasia, Sturge-Weber syndrome, Klippel-Trénaunay-Weber syndrome, Rendu-Osler-Weber syndrome, Wyburn-Mason syndrome, and Fabry's disease. For each condition, it describes the genetic basis, characteristic features, diagnostic criteria, clinical manifestations, treatment approaches, and











































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)


