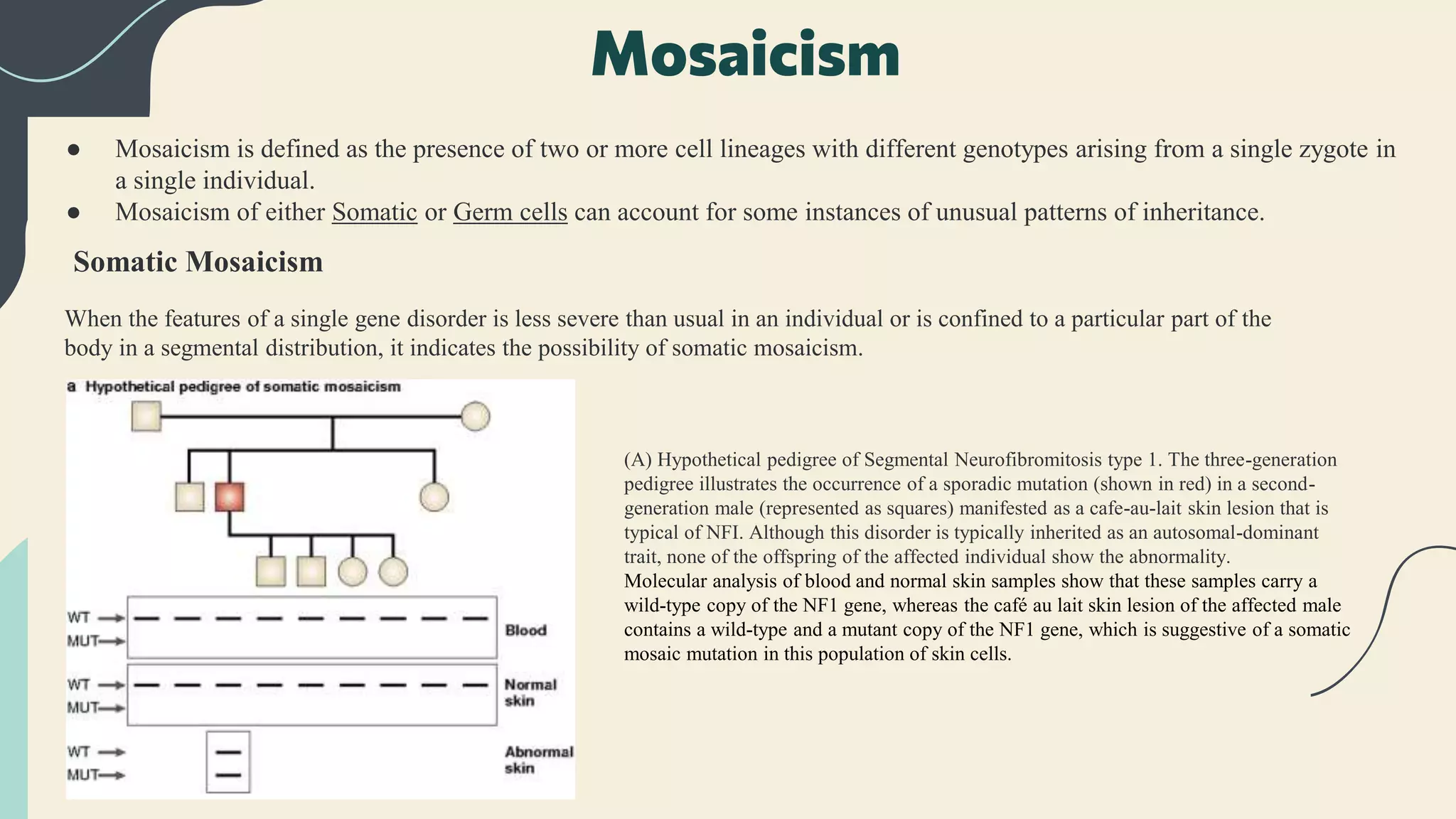
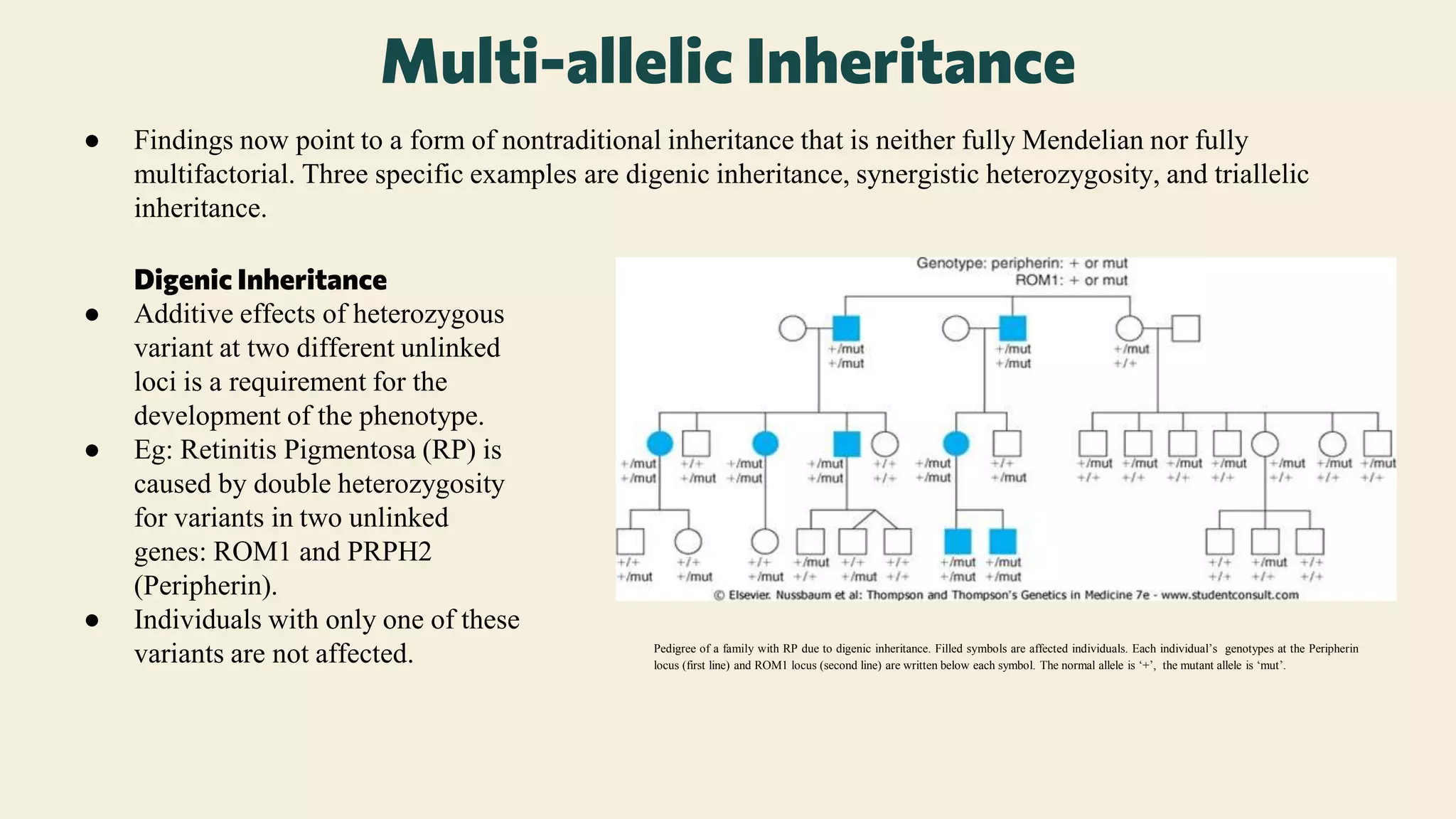
The document provides an overview of non-traditional inheritance mechanisms that deviate from Mendelian patterns, including triplet repeat expansions, genomic imprinting, and somatic mosaicism. It explains various genetic conditions, such as Fragile X syndrome and Prader-Willi syndrome, highlighting the complex interactions of genes and inheritance patterns that do not conform to classical inheritance laws. Additionally, it discusses concepts like uniparental disomy, mitochondrial inheritance, and multi-allelic inheritance, showcasing how these mechanisms contribute to the phenotypic expression of genetic disorders.