Download as PDF, PPTX

















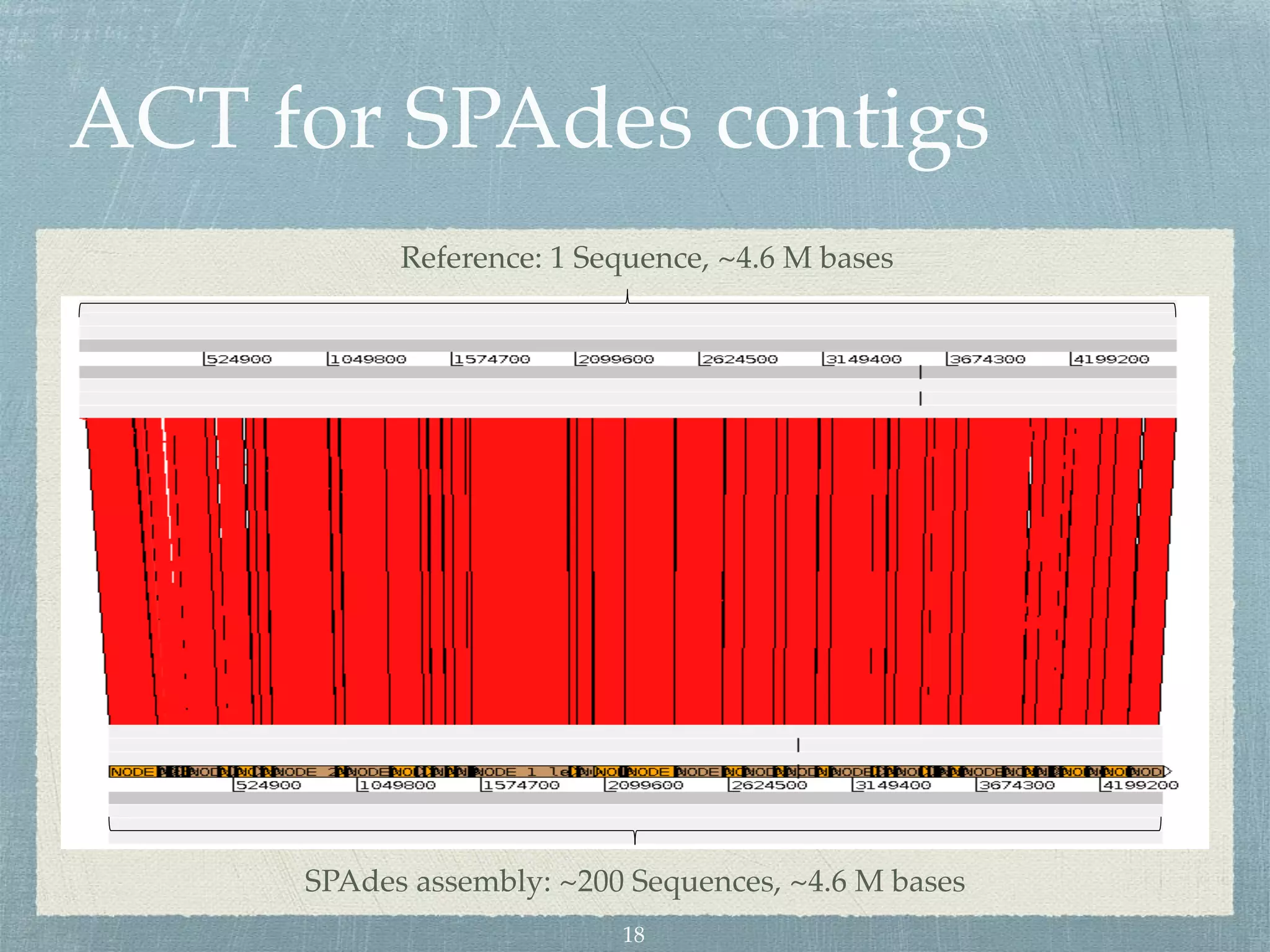



The document discusses different assembly strategies for Escherichia coli genome sequencing data, including assemblies generated from long reads only, short reads only, and a hybrid approach. It provides instructions for installing analysis tools and running assembly pipelines on the example data. Key findings are summarized, such as the MiniAsm assembly having lower accuracy but fewer contigs than SPAdes, and the hybrid assembly combining benefits of both approaches. Visualization of assembly alignments using ACT is demonstrated.



























































