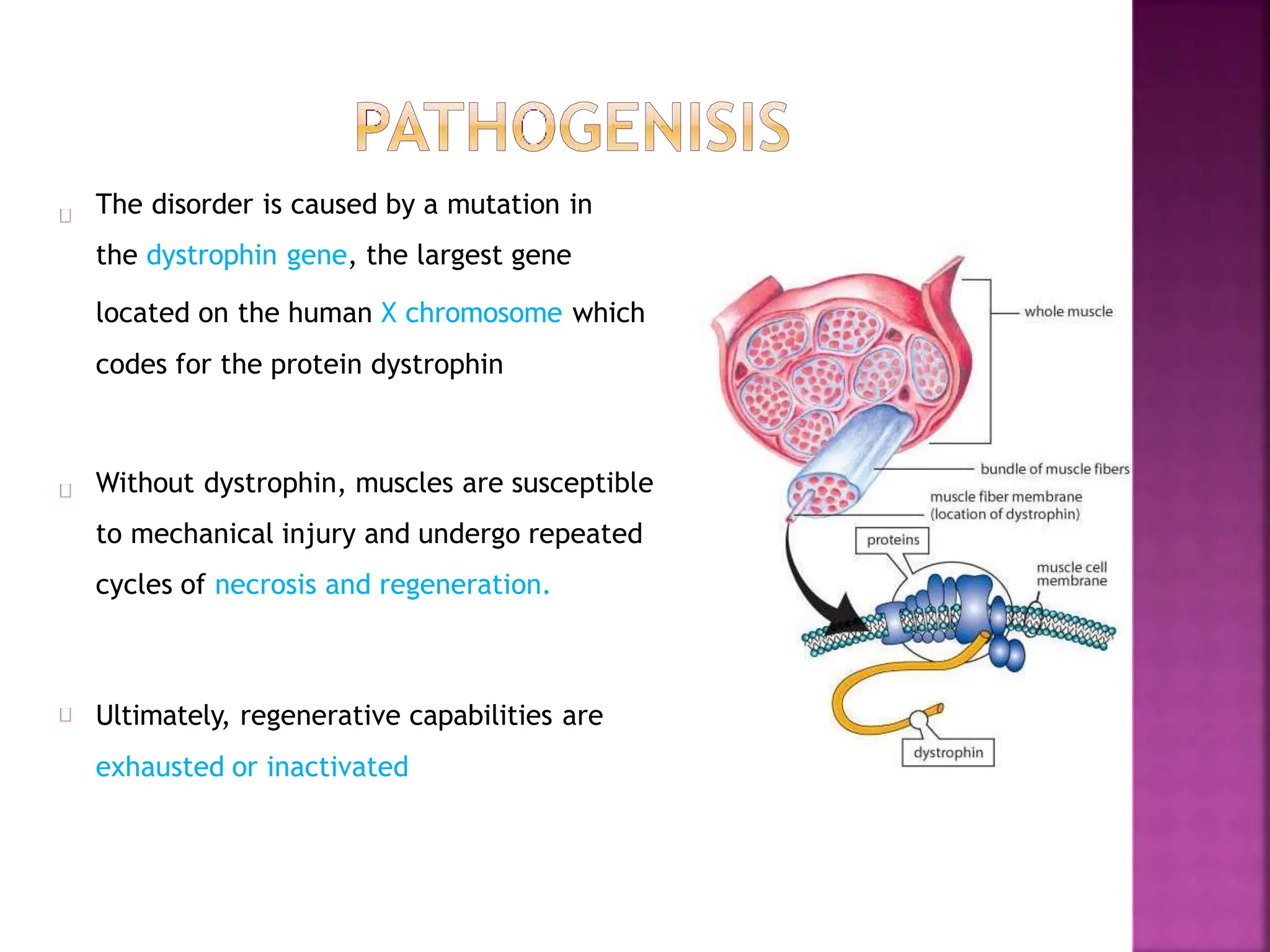
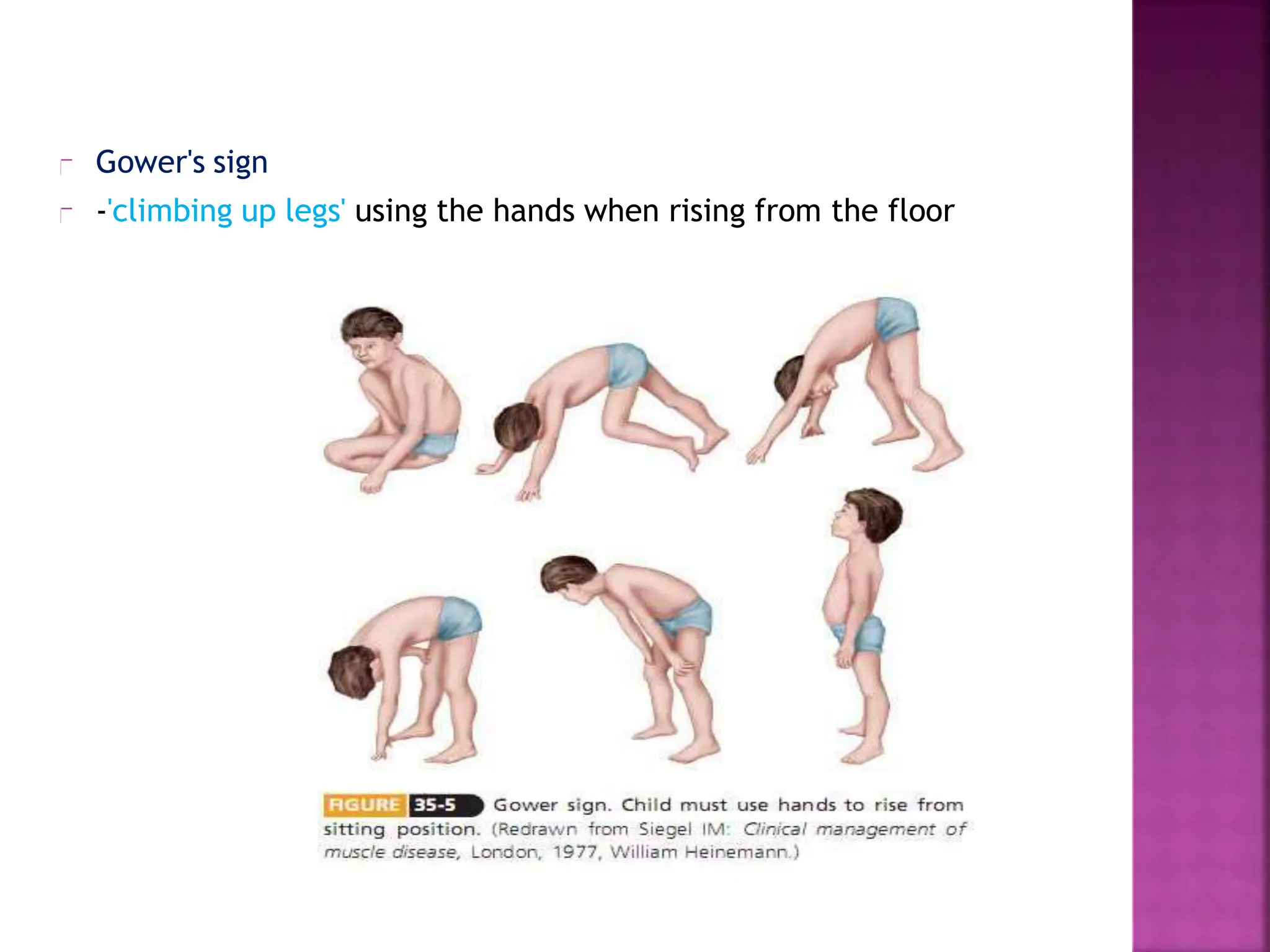
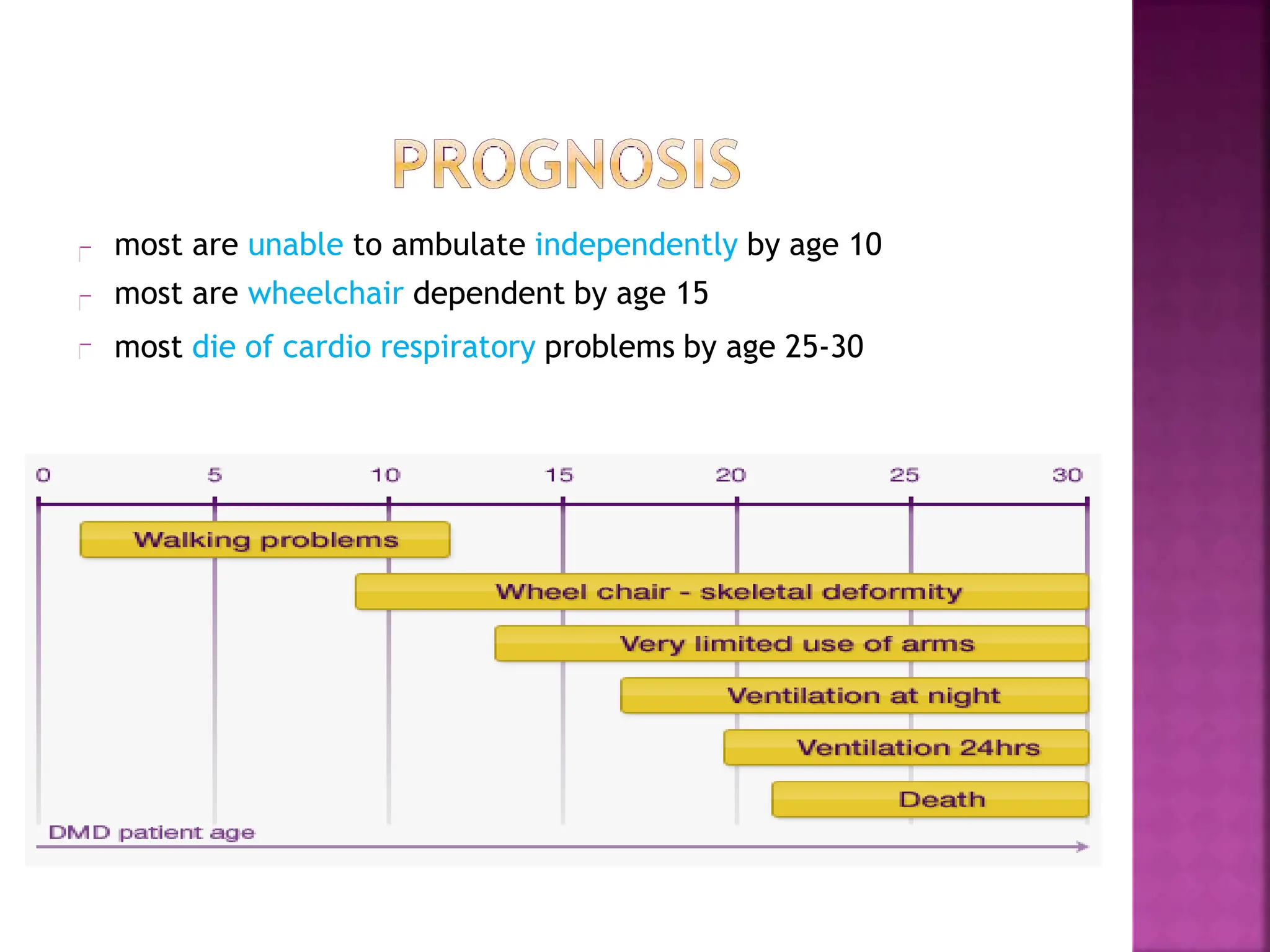
This document provides an overview of Duchenne muscular dystrophy (DMD). It begins with a brief history of the disease and its discovery. It then discusses the genetic basis and pathophysiology of DMD, which involves a mutation in the dystrophin gene. The clinical features and stages of DMD are outlined, from early ambulatory difficulties in childhood to loss of ambulation and eventual death often by the 30s due to respiratory or cardiac failure. Current treatment focuses on symptom management through physical therapy, bracing, medications, and nutritional support, though stem cell therapy and gene therapy show promise.