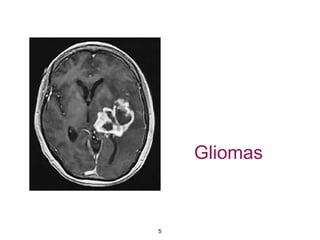
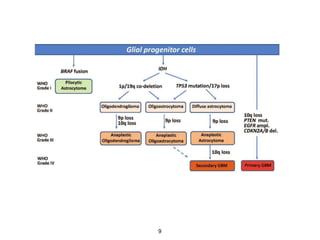
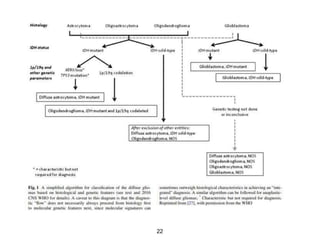
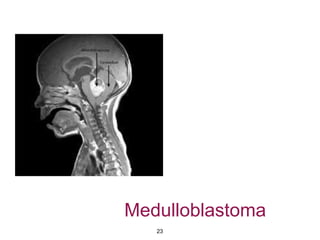
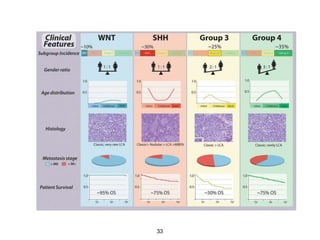
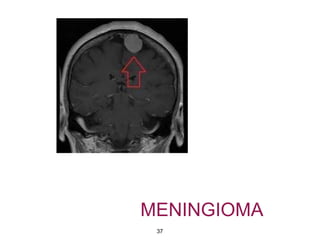
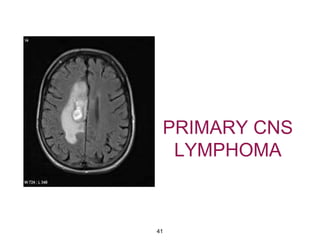
This document summarizes molecular studies in central nervous system (CNS) tumors. It discusses the need for molecular studies to integrate histologic and molecular data in tumor classification. It then reviews molecular markers for various CNS tumors including gliomas, medulloblastoma, ependymal tumors, and meningioma. For each tumor type, key genetic alterations are identified that have diagnostic, predictive or prognostic significance such as IDH1/2 mutations, 1p/19q codeletion, MGMT methylation, and others.































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)







![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)


