Downloaded 17 times









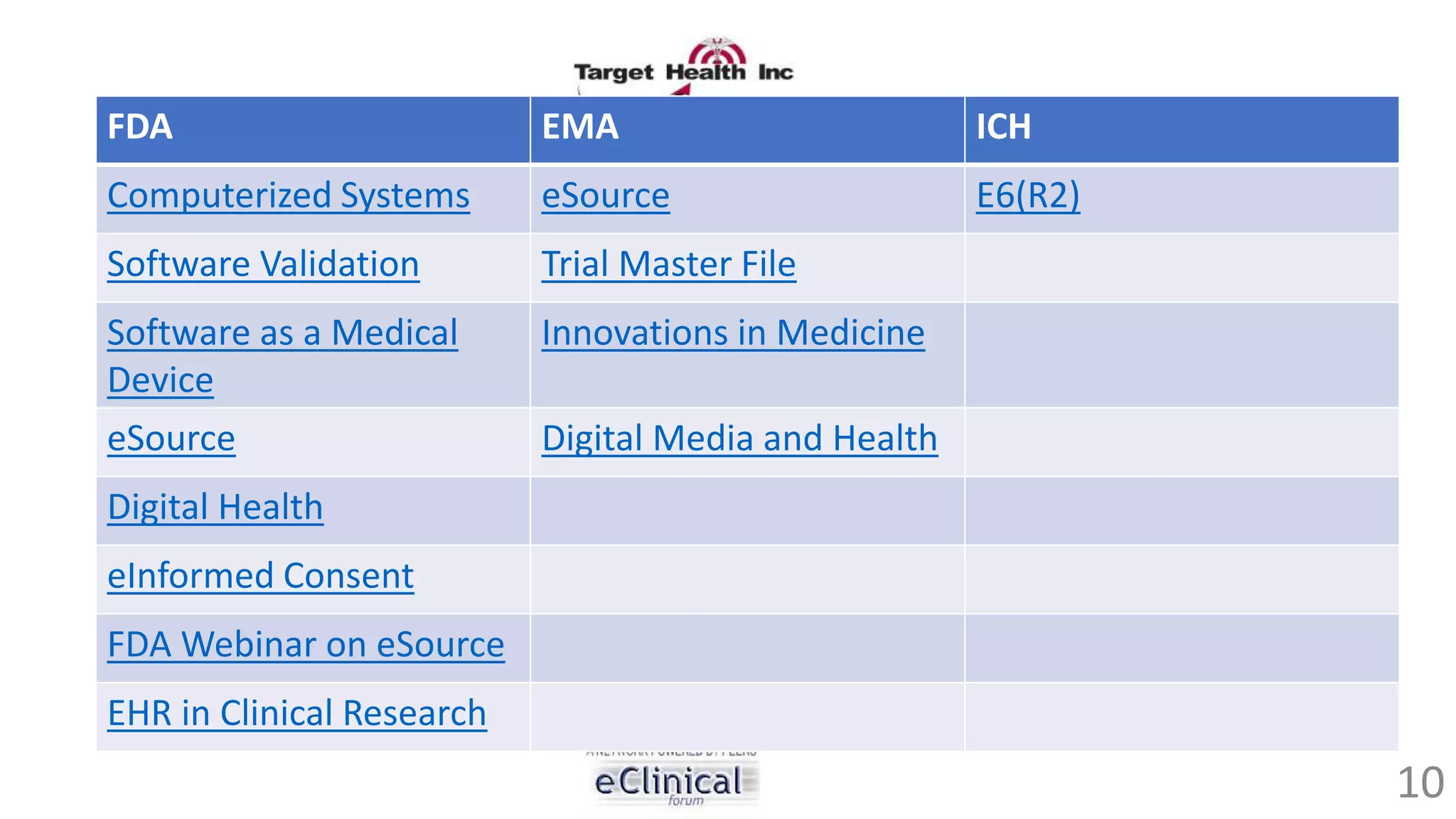

















The document discusses regulatory concerns and best practices for conducting virtual and paperless clinical trials, emphasizing the need for effective authentication and compliance with regulations such as 21 CFR Part 11. It highlights the role of technology, particularly mobile apps and electronic data systems, in improving data collection and reducing errors while ensuring patient safety and data integrity. The presentation also outlines necessary preparations for interactions with regulatory bodies and provides guidelines for ensuring that electronic systems are validated and used correctly.





































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)





