Downloaded 42 times

























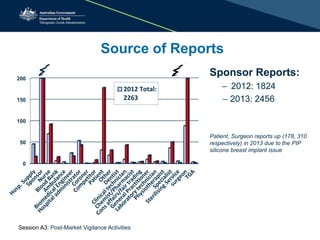
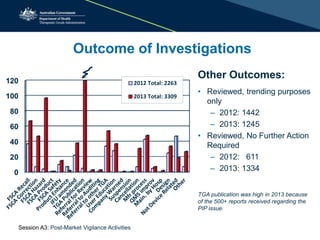


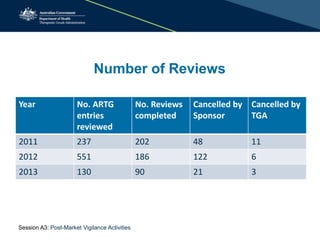



















The document outlines the role of the Therapeutic Goods Administration (TGA) in post-market vigilance and regulation of medical devices, emphasizing compliance monitoring, adverse event reporting, and safety assessments. It details the responsibilities of sponsors in reporting adverse events and highlights the procedures for investigating incidents and taking corrective actions. Additionally, it addresses the challenges in determining adverse events and the importance of thorough reporting to ensure patient safety.


























































































