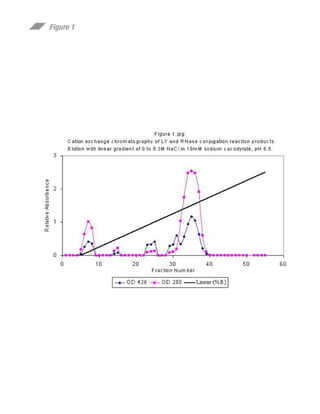
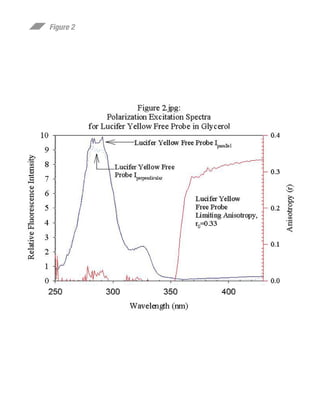
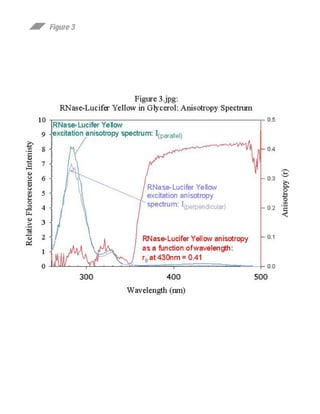
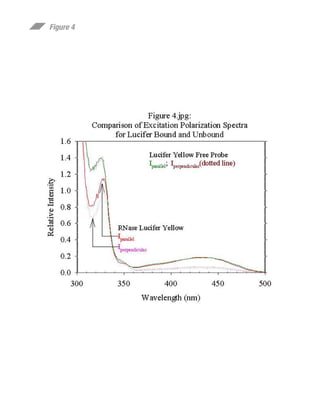
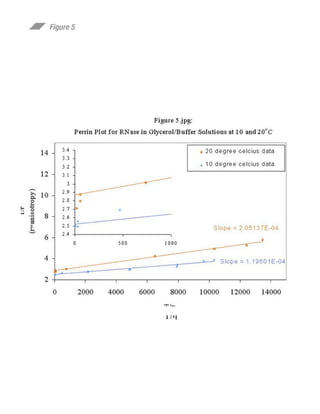
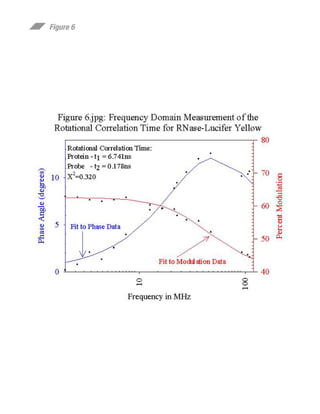
This document summarizes research on preparing and characterizing a conjugate of the protein ribonuclease A (RNase) with the fluorescent dye lucifer yellow (LY). The conjugate was prepared by covalently linking LY to an active site histidine residue on RNase. Steady-state fluorescence anisotropy measurements were performed on the conjugate at varying viscosities and temperatures. Time-resolved fluorescence measurements determined the rotational correlation time of RNase and the rigidly bound LY probe. The data suggest the fluorophore is rigidly bound to RNase at 10°C and can be used to study the rotational dynamics and self-assembly of RNase in solution.