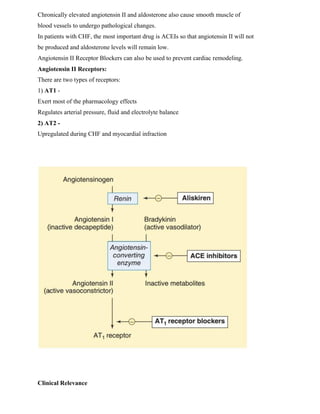
The Renin-Angiotensin-Aldosterone System (RAAS) is a hormone system that regulates blood pressure and fluid balance. It consists mainly of the hormones renin, angiotensin II, and aldosterone. Renin is released by the kidneys and cleaves angiotensinogen into angiotensin I. Angiotensin-converting enzyme then converts angiotensin I into the potent vasoconstrictor angiotensin II. Angiotensin II stimulates the release of aldosterone from the adrenal cortex and causes vasoconstriction, sodium retention, and increased blood pressure. Blocking this system is important for treating hypertension and other cardiovascular