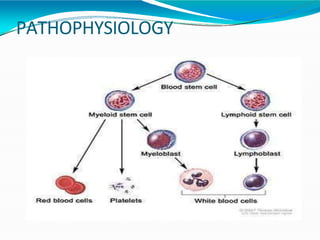
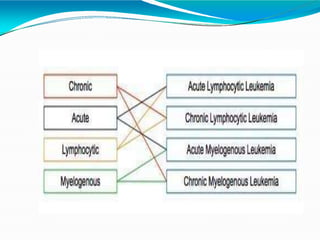
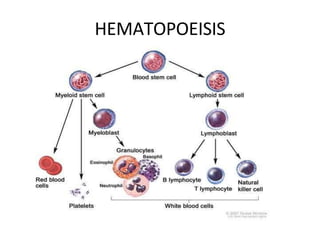
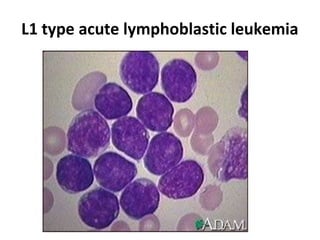
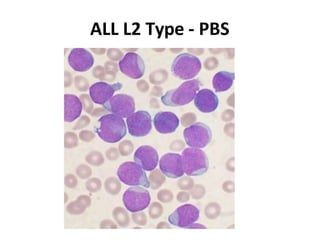
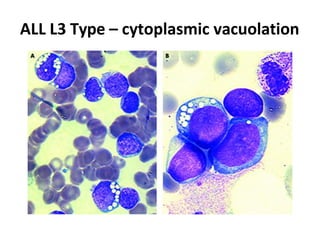
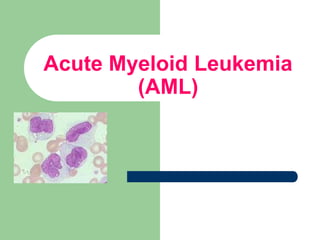
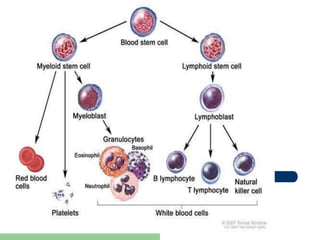
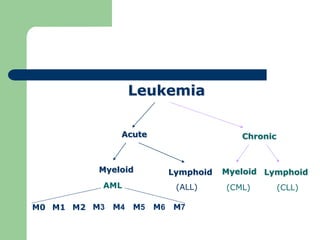
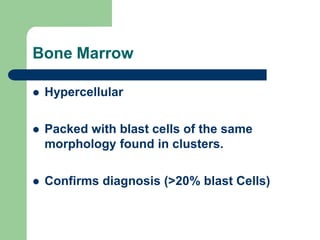
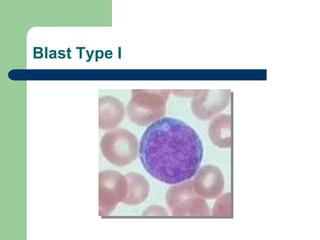
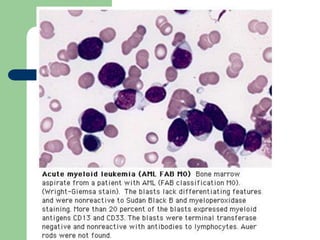
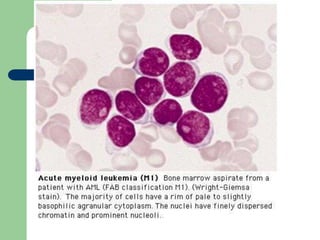
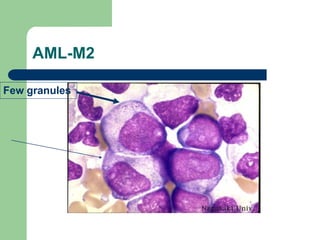
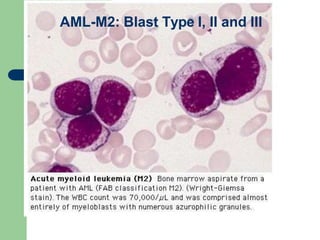
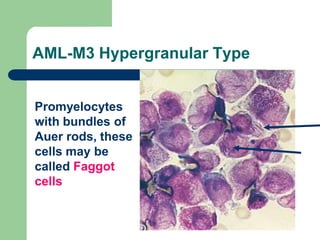
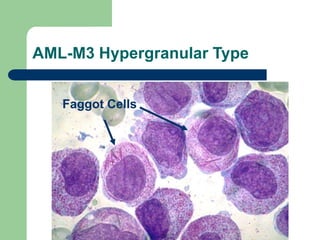
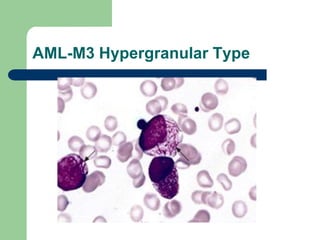
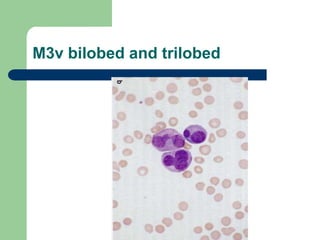
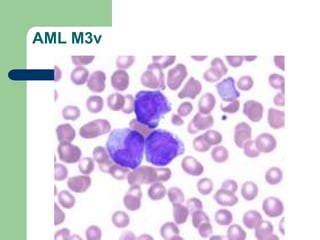
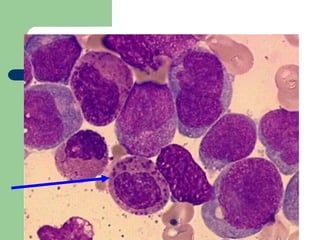
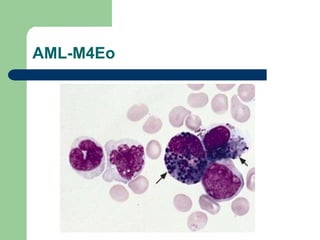
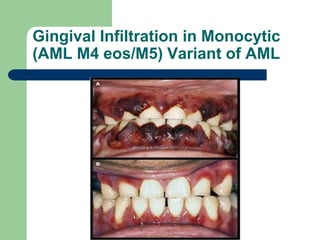
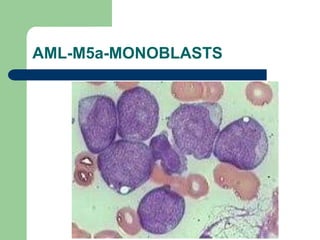
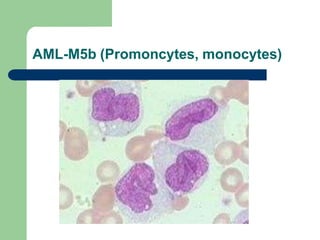
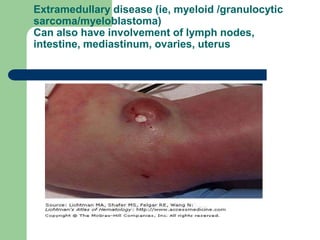
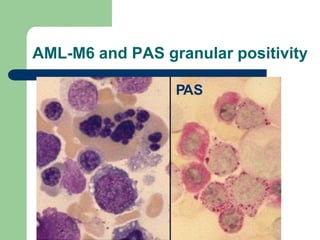
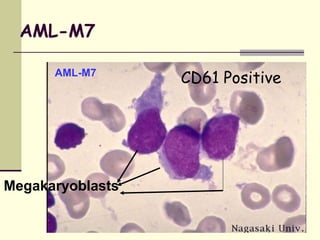
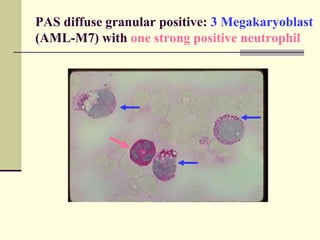
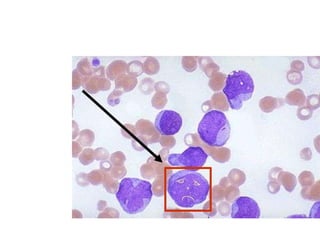
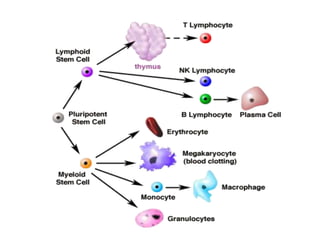
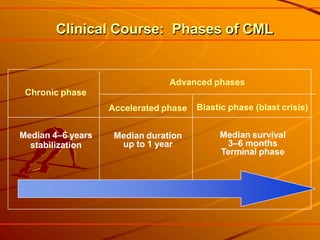
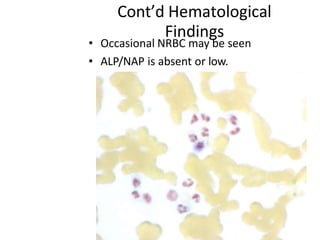
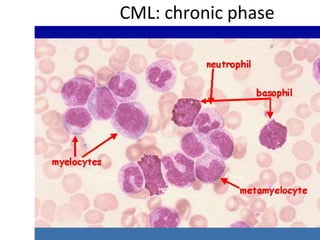
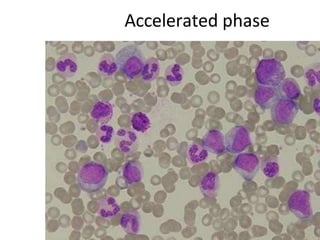
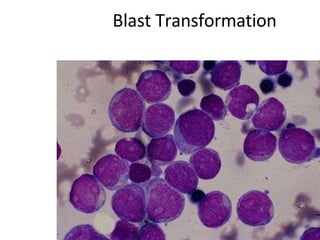
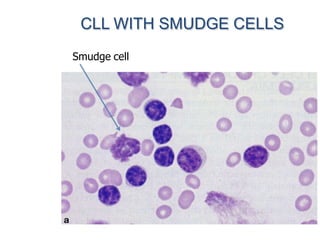
This document provides information on leukemia. It defines leukemia as a group of malignant disorders affecting the blood and bone marrow. The two main types are acute leukemias, including acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML), and chronic leukemias, including chronic myeloid leukemia (CML) and chronic lymphocytic leukemia (CLL). It discusses the etiology, pathophysiology, classification, and features of the different types of leukemia.