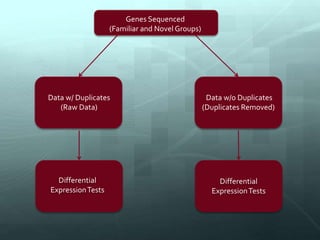
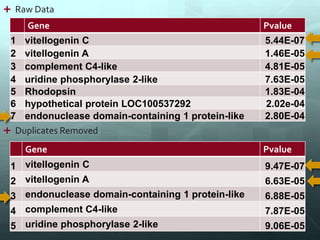
This document discusses the impact of removing exact duplicate reads from RNA-seq data on differential expression analysis results. The author conducted an experiment comparing differential expression analysis with and without duplicate removal on RNA-seq data from guppy brains. With duplicate removal, the total read count was lower but the top genes and p-values changed proportionally, suggesting duplicates were mainly PCR artifacts. The author concludes duplicate removal can change results quantitatively but not qualitatively, and future work could develop methods to eliminate PCR from sequencing.