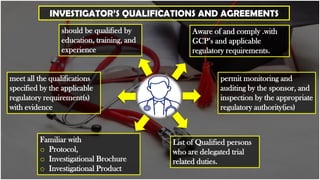
The document outlines the responsibilities of clinical trial investigators, including their qualifications, resource management, medical care for subjects, compliance with protocols, and safety reporting. Investigators must ensure informed consent, maintain accurate records, and communicate with regulatory bodies throughout the trial process. Additionally, it emphasizes the importance of adhering to good clinical practice (GCP) and regulatory requirements while protecting the rights and well-being of participants.





































![roles and responsibilities of Investigator[663]](https://cdn.slidesharecdn.com/ss_thumbnails/investigator663-210616055819-thumbnail.jpg?width=640&height=640&fit=bounds)

















![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)



































































