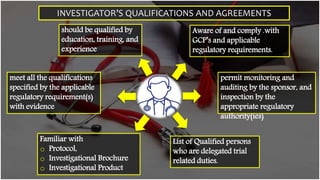
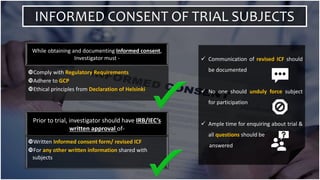
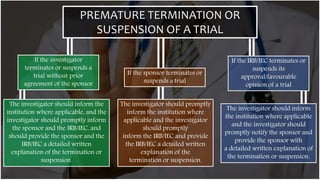
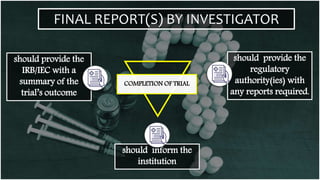
The document outlines the responsibilities and qualifications of clinical trial investigators, emphasizing the need for adequate resources, medical care for subjects, and compliance with regulatory protocols. It details requirements for informed consent, safety reporting, and communication with relevant authorities. The investigator must ensure adherence to Good Clinical Practice (GCP) and complete accurate records and reports throughout the trial process.












































![roles and responsibilities of Investigator[663]](https://cdn.slidesharecdn.com/ss_thumbnails/investigator663-210616055819-thumbnail.jpg?width=640&height=640&fit=bounds)


















































![Hypothalamus short notes on location, function and disorders by Dr. Neha [PT]...](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124142231-2b48143d-thumbnail.jpg?width=640&height=640&fit=bounds)




