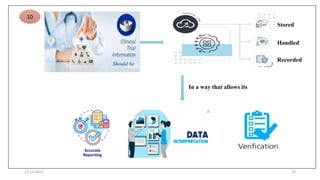
1. Good clinical practice (GCP) provides a standard to ensure clinical trials are scientifically valid and protect subject rights. GCP guidelines cover trial conduct, monitoring, records, and reporting.
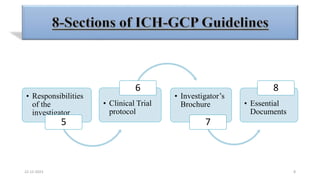
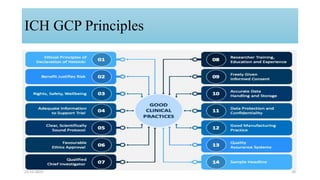
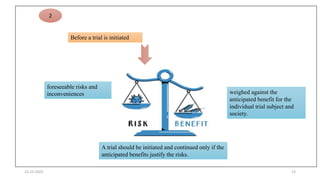
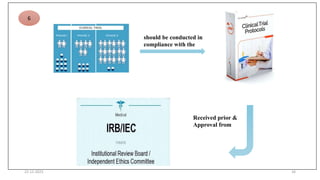
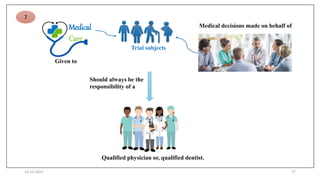
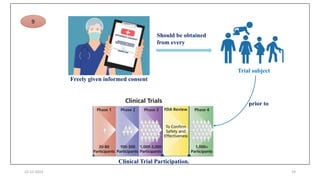
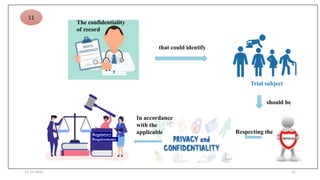
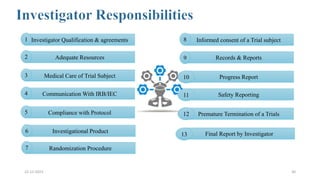
2. Ethics committees review protocols to ensure trials are ethical and risks are outweighed by benefits. Investigators must be qualified and obtain informed consent. Sponsors design and fund trials and ensure product quality.
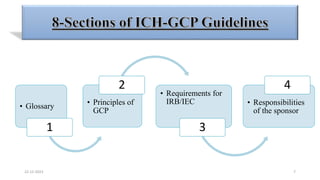
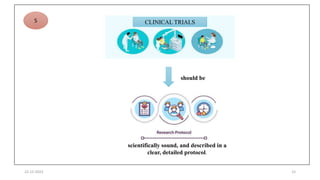
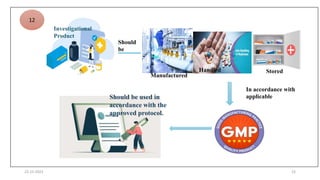
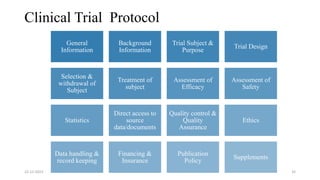
3. Clinical trial protocols describe trial objectives, methodology, and organization. Investigator brochures provide information on investigational products. Essential documents for GCP compliance include the protocol, consent forms, product information, records, and reports.



































![Volume 9 A Guidelines On Pharmacovigilance[1]](https://cdn.slidesharecdn.com/ss_thumbnails/volume9aguidelinesonpharmacovigilance1-12816046179281-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)




![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)













































