Downloaded 40 times































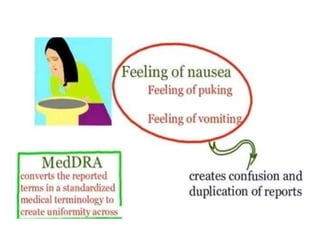

















































![e)Data processing
Data is best managed electronically by computer,
wherein, data is entered in a hierarchical format according
to product name, medicine name or therapeutic category.
This facilitates recording detailed case information and
easy retrieval Internationally accepted terminologies
regarding classification of medicines (Anatomical
Therapeutic Chemical [ATC], International Nonproprietary
Names [INN]) and ADRs e.g. WHO Adverse Reaction
Terminology (WHO ART), Medical Dictionary for
Regulatory Activity (MedDRA) should be used, so that the
data can be globally shared.](https://image.slidesharecdn.com/informationonpharmacovigilance-230310074054-6ccd42af/85/Information-on-Pharmacovigilance-pptx-83-320.jpg)



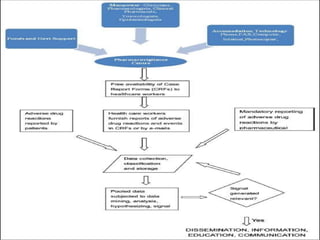
















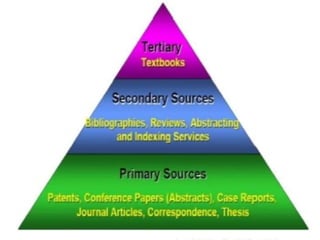
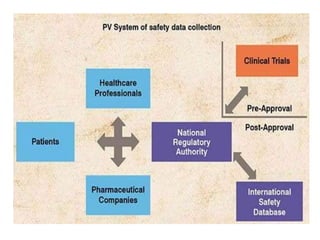
This document provides information on establishing pharmacovigilance programs and resources in pharmacovigilance. It discusses the importance of classification systems like ATC, ICD, and INN for monitoring drugs and adverse events. Primary resources include clinical trial data and reports, while secondary resources summarize primary sources. Tertiary sources compile information from multiple sources. Establishing an effective pharmacovigilance program requires a structured organization, trained staff, coordination between stakeholders, and support from regulatory authorities and healthcare priorities. Monitoring drug safety is crucial for public health.









































































