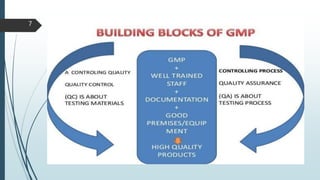
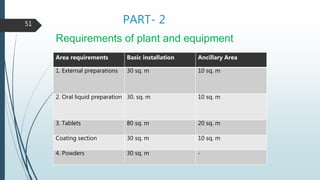
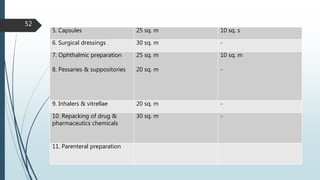
This document discusses Good Manufacturing Practices (GMP) for pharmaceutical manufacturing. It begins with an overview of why GMP is important for ensuring consistent quality and safety of medicines. It then outlines the key principles of GMP, including requirements for facilities, equipment, documentation, personnel, sanitation, and quality control. The document provides details on specific GMP requirements for premises, materials, production areas, equipment, packaging, batch records, and quality assurance systems. It also discusses self-inspection and quality auditing.