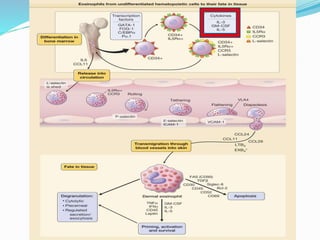
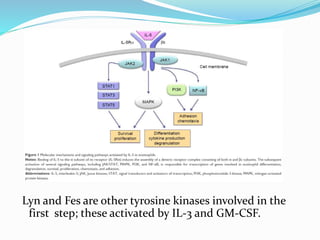
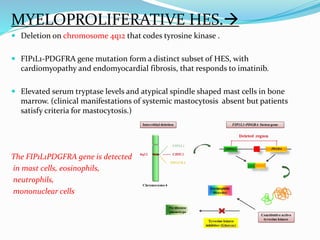
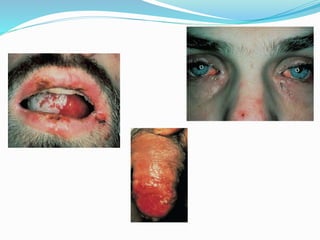
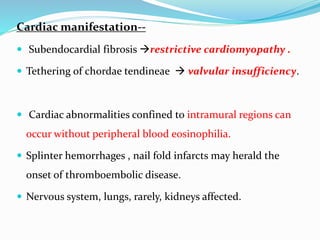
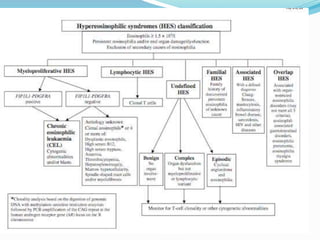
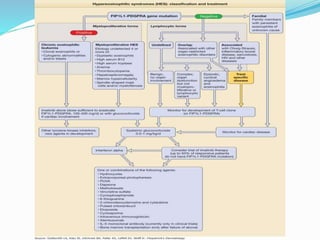
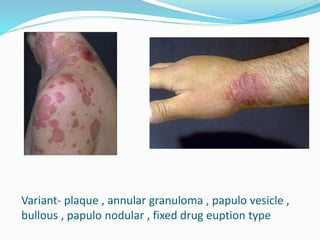
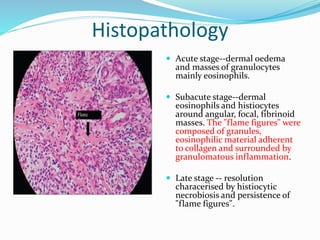
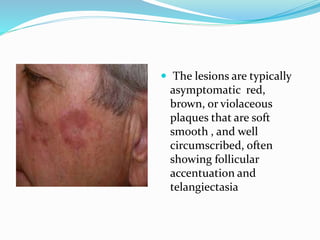
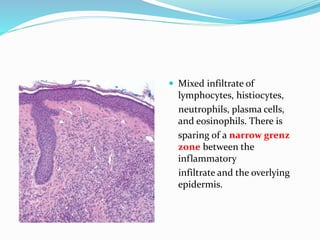
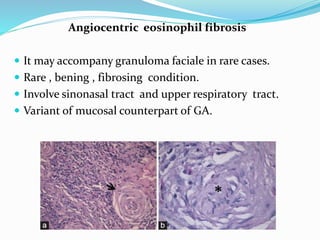
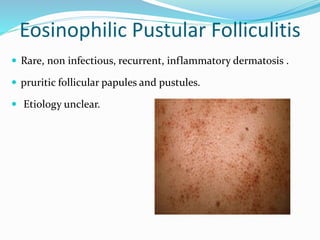
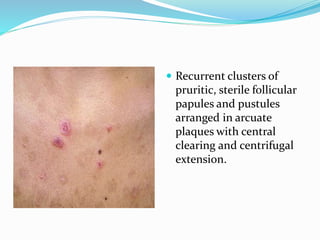
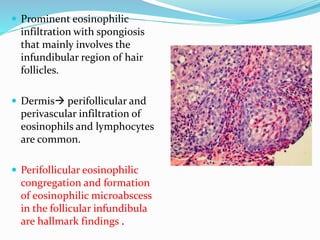
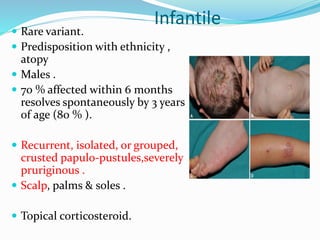
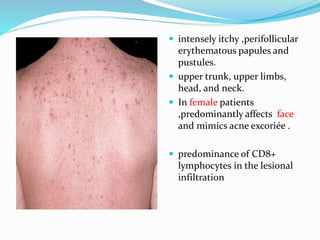
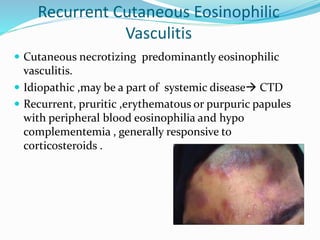
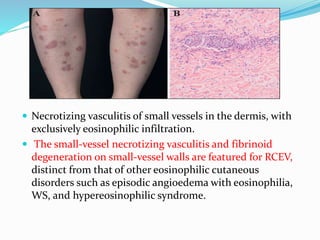
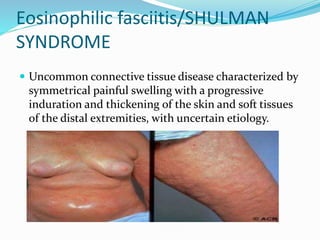
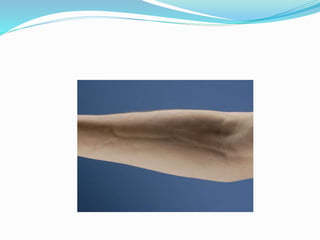
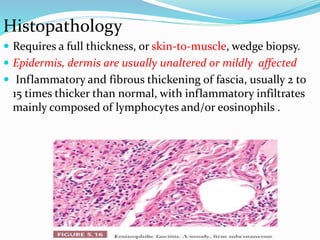
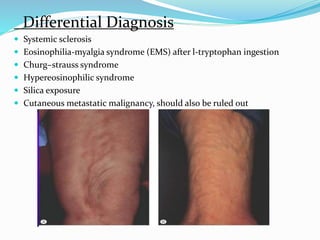
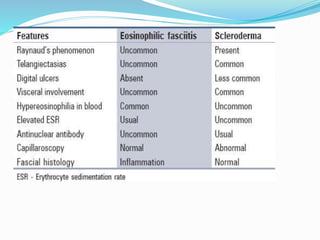
This document provides information on eosinophils and various eosinophilic conditions. It begins with basic facts about eosinophils as bone marrow-derived cells that normally make up 6% of white blood cells. It then discusses eosinophilic syndromes like hypereosinophilic syndrome and its subtypes. Other conditions discussed in detail include Well's syndrome, eosinophilic cellulitis, lymphocytic variant HES, myeloproliferative HES, granuloma faciale, erythema elevatum diutinum, eosinophilic pustular folliculitis, and recurrent cutaneous eosinophilic vasculitis. Clinical findings, histopathology, differential diagnoses