Download to read offline




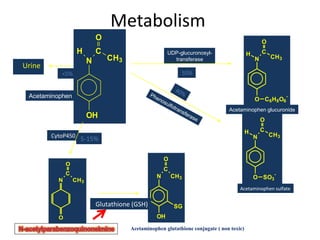

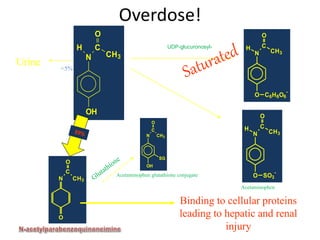
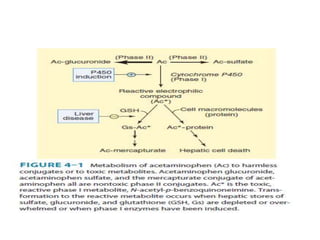






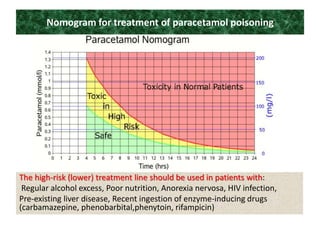






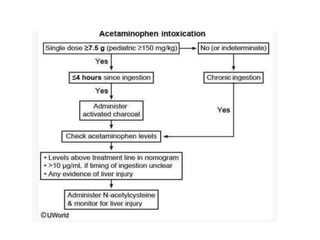





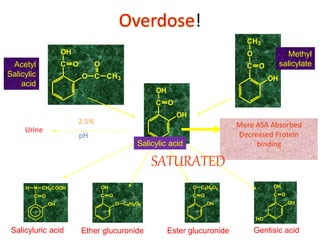



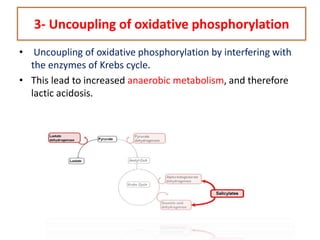

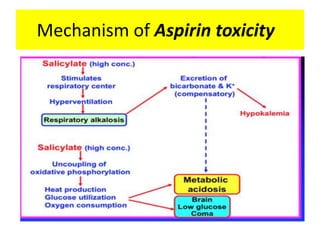












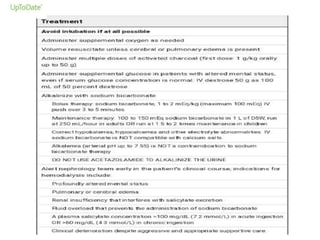




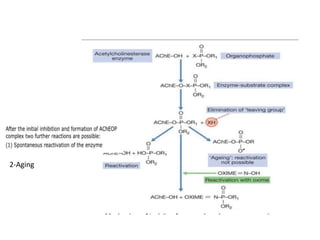





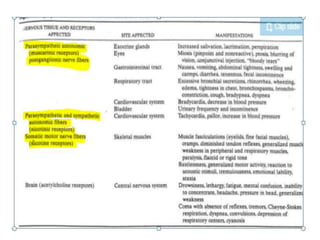





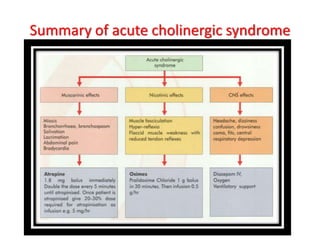



Poisoning is commonly accidental or suicidal and occurs through substances in homes or workplaces. Acetaminophen is a common oral analgesic and antipyretic that is generally safe in proper doses but can cause liver toxicity and failure in overdose due to formation of a toxic metabolite. Clinical features progress from nausea and vomiting to liver damage and failure over 72-96 hours. Treatment involves assessing for toxicity using acetaminophen levels and the Rumack-Matthew nomogram to determine if N-acetylcysteine is needed. Organophosphates inhibit acetylcholinesterase, accumulating acetylcholine and causing nicotinic and muscarinic effects like increased secretions, nausea, weakness and seizures. Salicy



































































