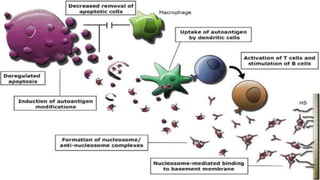
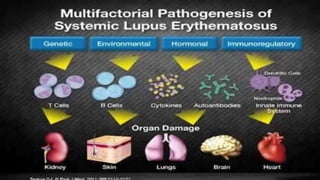
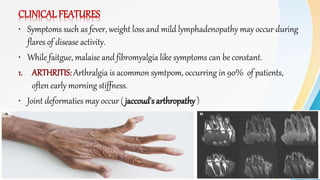
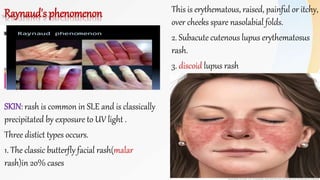
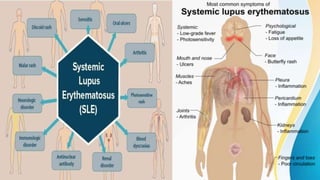
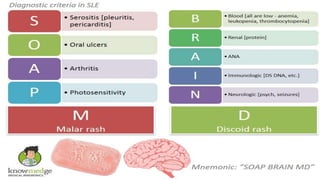
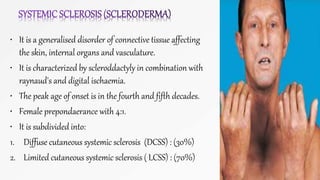
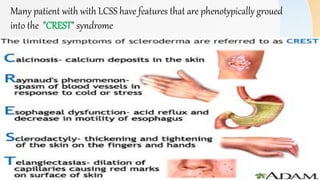
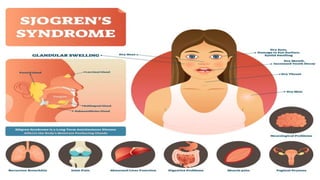
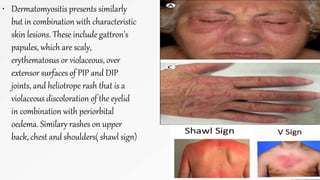
Connective tissue diseases share features of immune dysregulation and autoantibody production directed at nuclear components, causing widespread tissue damage. Systemic lupus erythematosus is characterized by arthritis, rashes, kidney involvement and positive ANA and anti-dsDNA antibodies. Systemic sclerosis involves skin thickening from fibrosis, Raynaud's phenomenon, and autoantibodies like anti-Scl-70. Polymyositis and dermatomyositis cause proximal muscle weakness and inflammation with skin lesions in dermatomyositis.