Downloaded 32 times











!["FORM 5-F"
[See rule 26 (l)]
Common Technical Document (CTD) for Registration of Human Drugs
contents o fM0dule 1 :Admtntstrattve part
HeadingSection Sub-
Section
1.1
1.2
1.3
1.3.1
Covering Letter and Fee Deposit Slip
Table of Contents (From Module 1 to Module 5)
Applicant Information
Name, address and contact details of Applicant I Marketing Authorization
Holder:
Name, address and contact details of Manufacturing site. Specify
whether the Applicant is:
1.3.2
1.3.3
a.
b.
c.
Valid
D Manufacturer
D Importer
Dls involved in none ofthe above (contract giver)
Drug Manufacturing License (DML) of manufacturer I Applicant or1.3.4
Drug Sale License, whichever is aoolicable.
Evidence of approval of manufacturing facility I Approved Section from
Licensing Authority
List of already approved registered drugs in this section
1.3.5
1.3.6
1.3.7 Identification of Signature(s) of authorized persons, Incharge
Quality Control and Incharge Quality Assurance
Manufacturer's Site Master File and Credential (for importer)
Type ofAoolication
Application is for the registration of:
D New Drug Product (NDP)
D Generic Drug Product (GDP) Pharmaceutical
product is intended for: D Domestic sale
D Export sale
D Domestic and Export sales
For imported products, please specify one of following: D
Finished Pharmaceutical Product Import
D Bulk Import and local repacking (specify status of bulk)
D Bulk Import Local Repacking for Export purpose only
Production,
1.3.8
1.4
1.4.1
1.4.1
1.4.2](https://image.slidesharecdn.com/ctdpakistan-181014100423/85/Common-Tecnical-Dossier-CTD-implementtion-in-Pakistan-12-320.jpg)




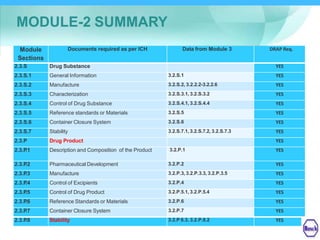

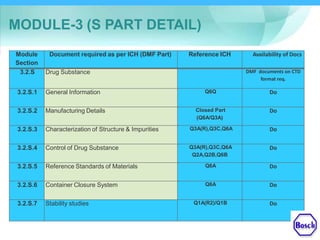
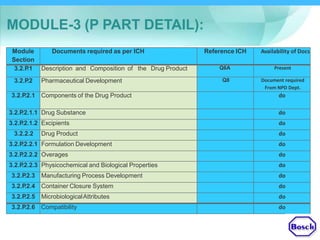





The document discusses DRAP's implementation of the Common Technical Document (CTD) format as the standard dossier for drug product registration applications in Pakistan beginning January 1, 2019. The CTD format was developed by international regulatory agencies to harmonize technical documents for new drug approvals. It aims to standardize information and reduce delays in the review process. The document outlines the history and purpose of the CTD, as well as the regions that have adopted it. It provides an overview of the five CTD modules and the key administrative information required in Module 1 for DRAP registration applications.






































![cmc [ chemistry manufacturing control ]](https://cdn.slidesharecdn.com/ss_thumbnails/presentation2222ra-181120122336-thumbnail.jpg?width=640&height=640&fit=bounds)




























