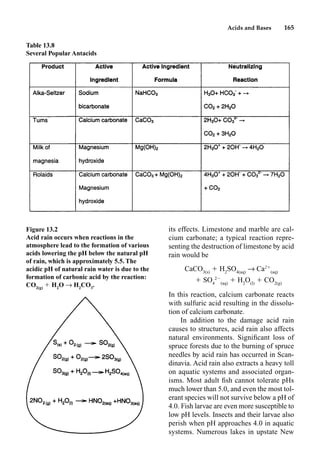
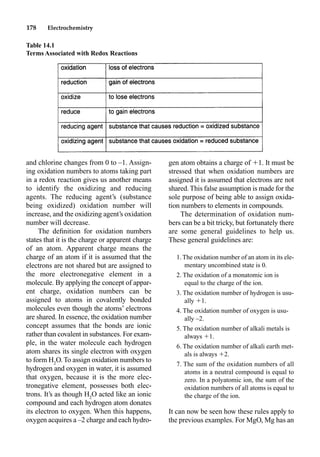
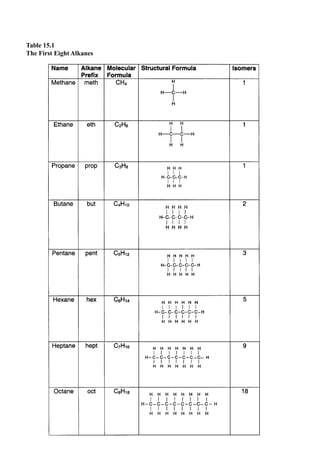
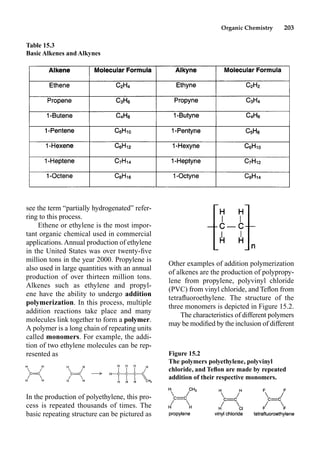
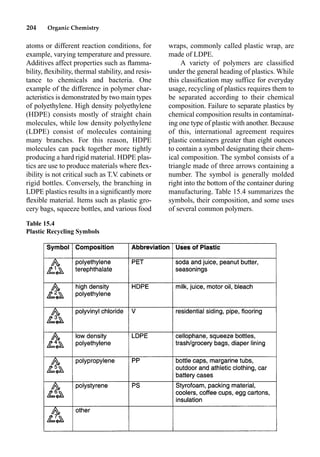
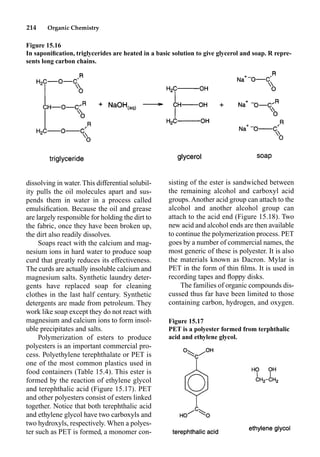
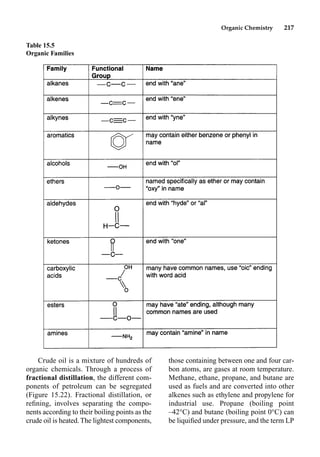
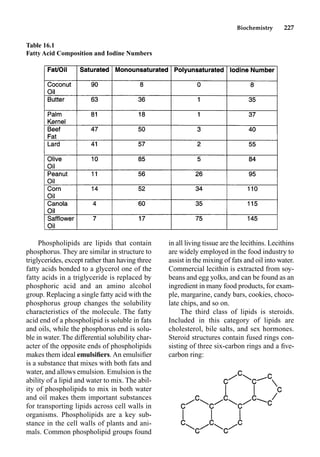
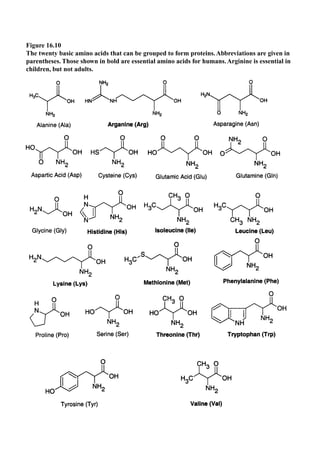
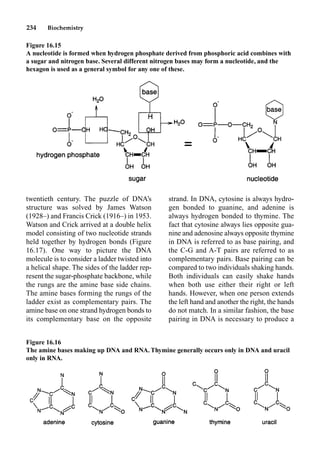
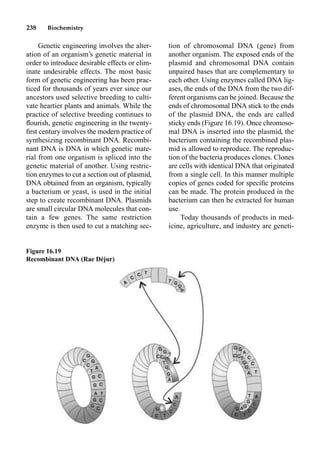
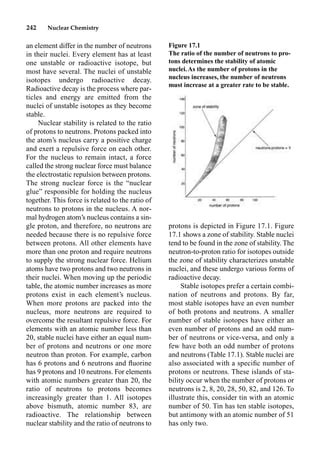
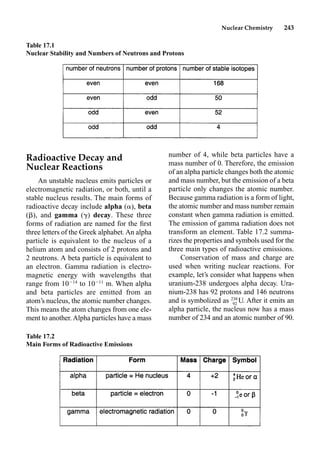
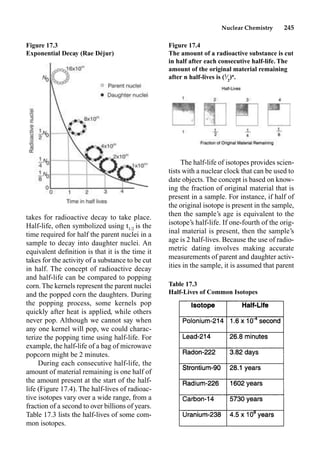
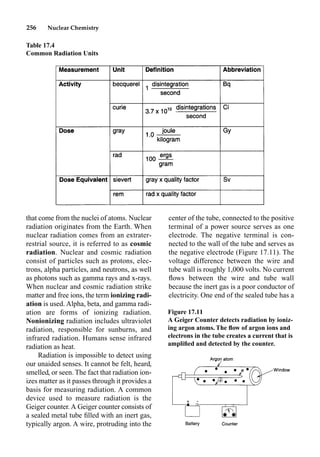
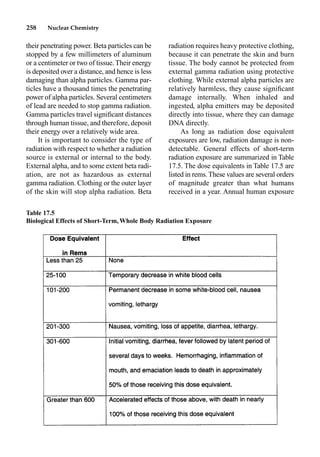
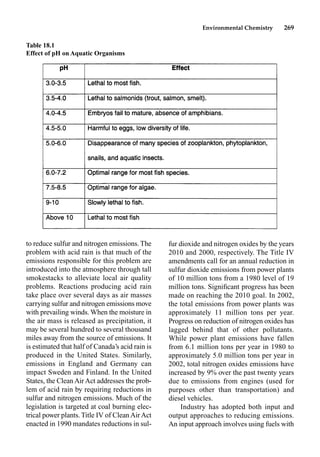
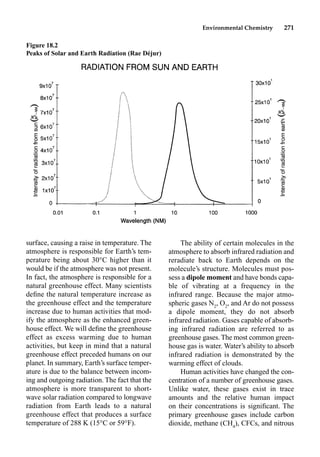
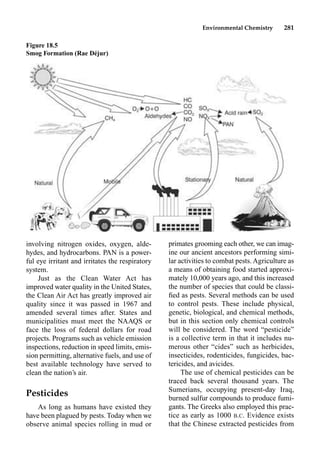
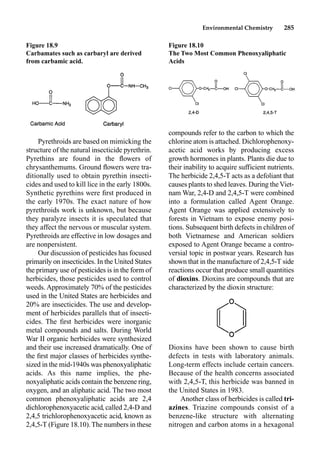
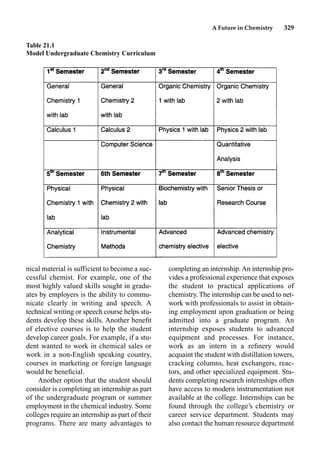
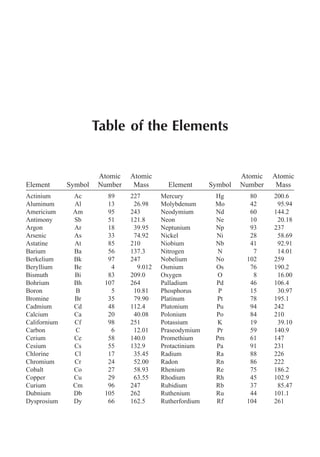
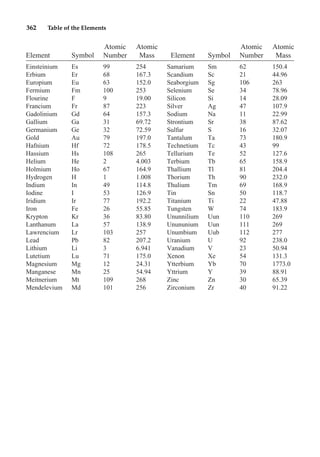
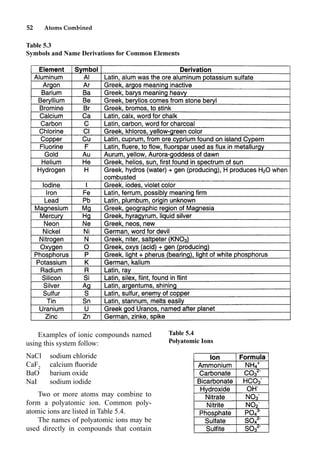
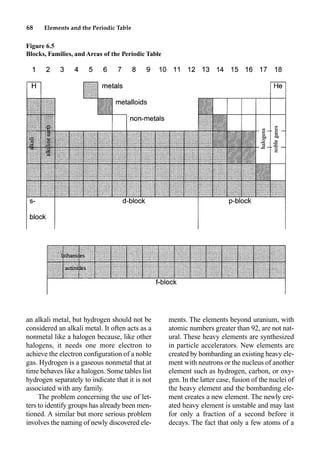
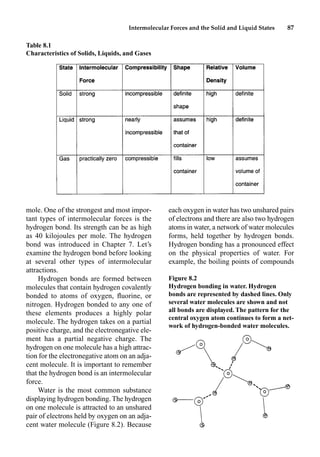
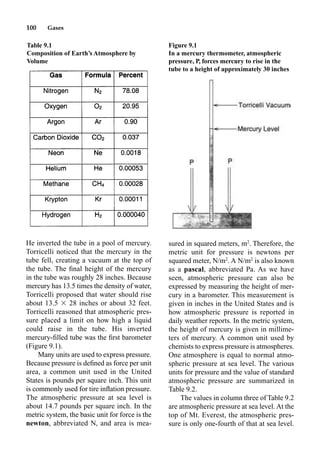
This document provides an overview and summary of the book "The Basics of Chemistry" by Richard Myers. It was published in 2003 by Greenwood Press as part of their "Basics of the Hard Sciences" series, edited by Robert E. Krebs. The book aims to introduce high school and college students to basic chemistry concepts in an accessible way. It covers a wide range of topics from the history of chemistry to modern subdisciplines like organic chemistry, biochemistry, and environmental chemistry. The book also discusses careers in chemistry and includes experiments and activities for students.


















































































































































![Kinetics and Equilibrium 151
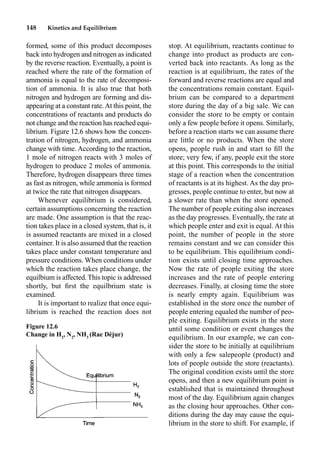
The final stress to be considered is a
change in temperature. To apply Le Châte-
lier’s Principle with temperature changes,
the sign of H for the reaction needs to be
known. The H in our example is 131
kilojoules. This indicates that the forward
reaction is endothermic and the reverse
reaction is exothermic. When the tempera-
ture of a system at equilibrium is increased,
the equilibrium will favor the endothermic
reaction. One way to think of the effect of
temperature is to think of energy as a reac-
tant or product. This is seen when the for-
ward and reverse reactions are written as
two separate reactions:
Forward Reaction
131 kJ C(s)
H2
O(g)
p CO(g)
H2(g)
Endothermic
Reverse Reaction
CO(g)
H2(g)
p C(s)
H2
O(g)
131 kJ
Exothermic
In an endothermic reaction, energy can be
considered a reactant, and in an exothermic
reaction, energy can be considered a prod-
uct. When the temperature of a system at
equilibrium is increased, energy flows into
the system. To relieve this stress, energy is
used in the endothermic reaction to estab-
lish a new equilibrium. The effect of adding
energy is no different from the effect of
adding another reactant, for example, steam.
Decreasing the temperature of the system
removes energy from the system; to coun-
teract this stress, the exothermic reaction is
favored.
The Equilibrium Constant
Two Norwegians named Cato Maximil-
ian Guldberg (1836–1902) and Peter Waage
(1833–1900) carried out a comprehensive
study of equilibrium between 1864 and
1879. Their work led to what is known as
the law of mass action. For a general
reversible reaction given by the equation: aA
bB m cC dD, the law of mass action
states an equilibrium constant, Keq
, can be
defined at a particular temperature:
The small letters are the coefficients in the
balanced equation and the brackets indicate
the molar concentrations (moles per liter) of
the reactants and products. Applying the law
of mass action to the hydrogen gas equation
gives
Notice that solid carbon does not appear in
the reaction. Again, this is because the con-
centration of solid carbon is constant and is
assumed to be part of Keq
. The value for Keq
is approximately 2 at 1,000°C.
Values for equilibrium constants have
been tabulated for numerous reactions
occurring at various temperatures. Because
many of these constants are associated with
industrial processes, the temperature at
which a Keq
is reported varies considerably.
That is, it might seem odd that a Keq
value
for a reaction is given at what seems like an
arbitrary temperature, but it should be
remembered that these constants have prob-
ably been determined with regard to a spe-
cific industrial process, for example,
1,000°C for the production of hydrogen gas
from steam and coke (carbon).
Values for equilibrium constants vary
widely. For example, consider the following
two reactions:
N2(g)
O2(g)
m 2NO(g)
Keq
1.0 1031
@ 25°C
H2(g)
Cl2(g)
m 2HCl(g)
Keq
1.8 1033
@ 25°C
c d
[C] [D]
K ⳱
eq a b
[A] [B]
[CO][H ]
2
K ⳱
eq
[H O]
2](https://image.slidesharecdn.com/jsvnpjstgguna8iuxpba-chemistry-221009154822-adb167b6/85/chemistry-pdf-165-320.jpg)