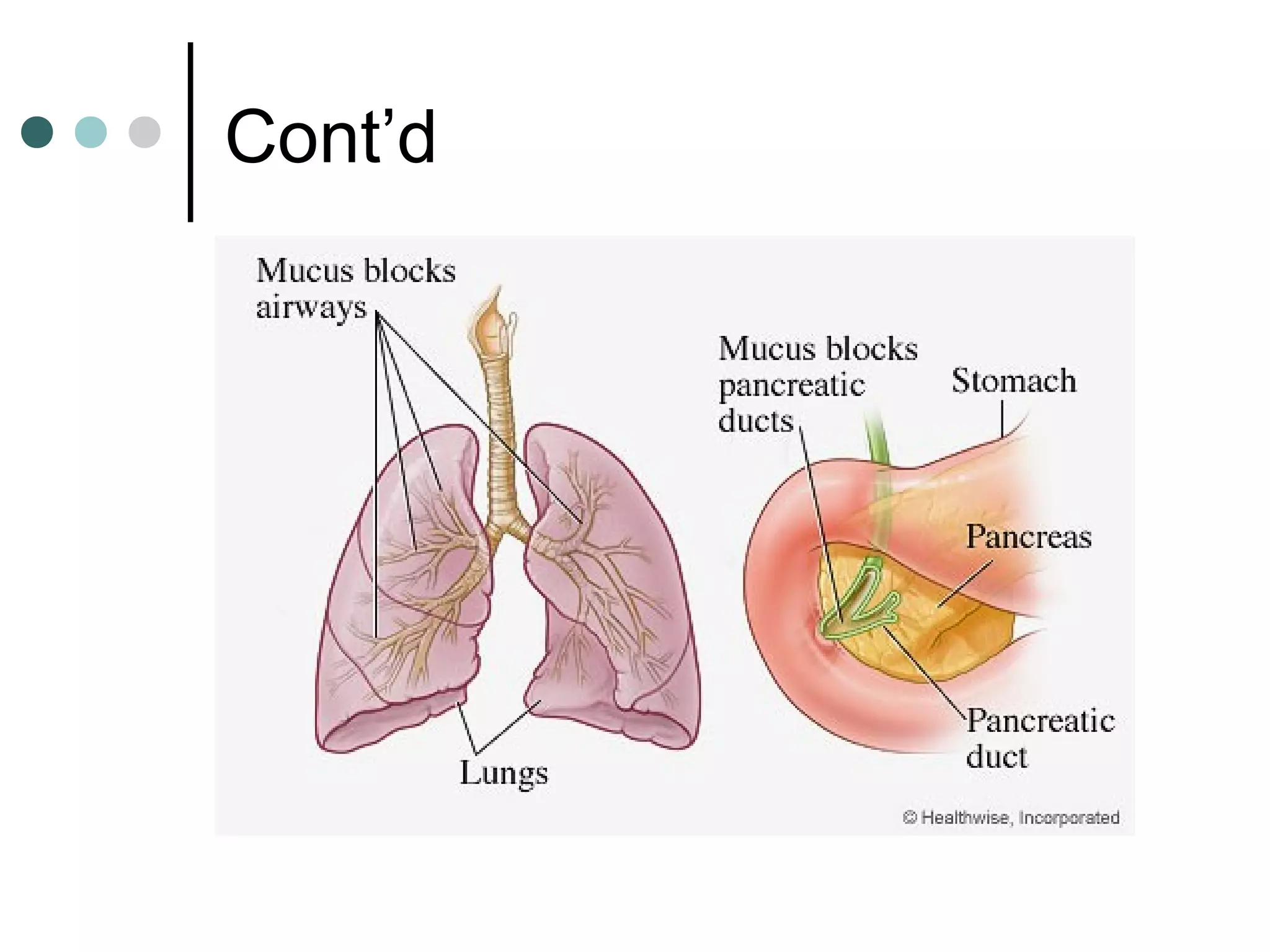
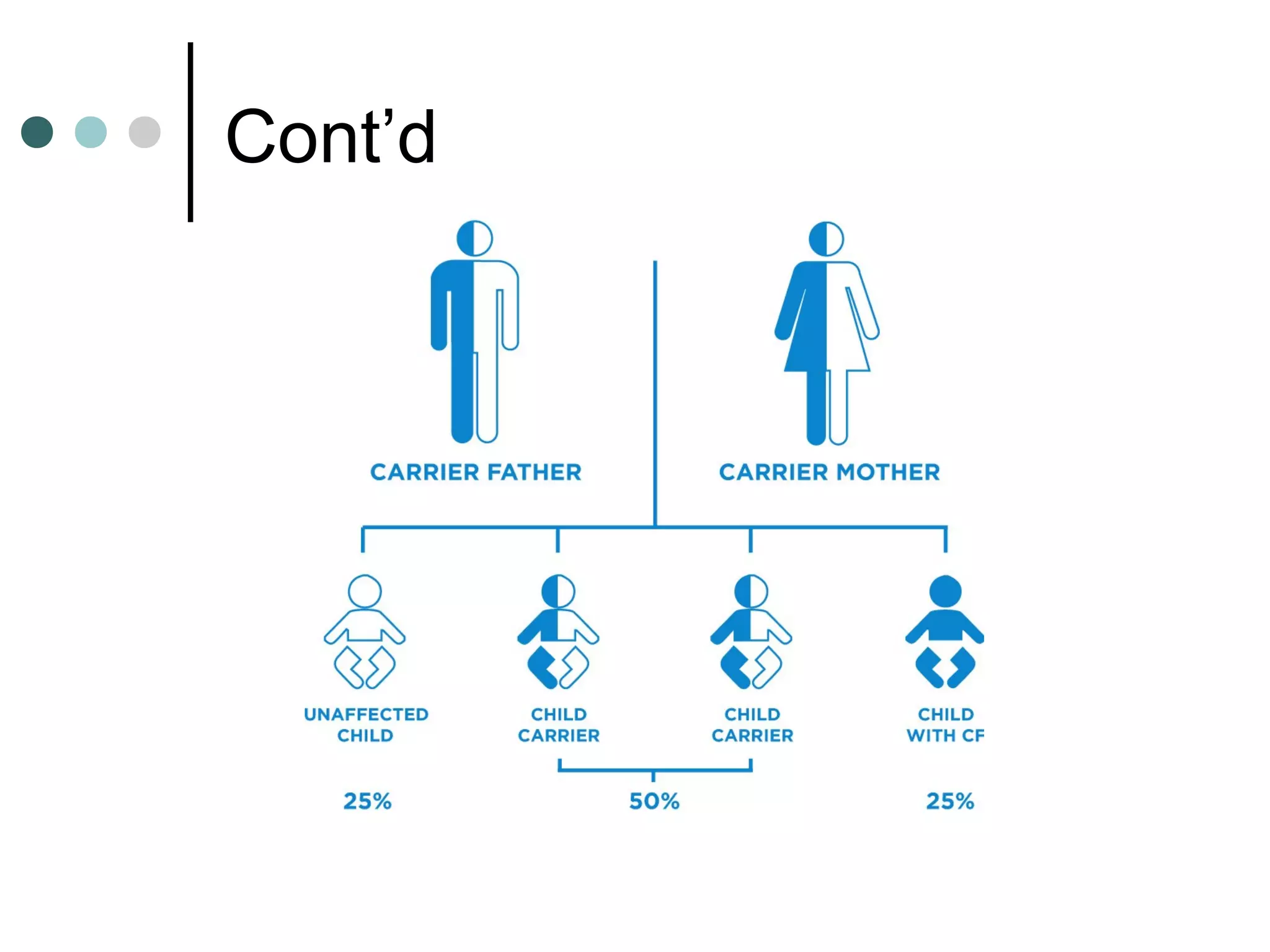
Cystic fibrosis is a genetic disease that causes thick, sticky mucus to build up in the lungs and digestive system. It is caused by inheriting two copies of a defective gene, one from each parent. Symptoms vary but often include salty skin, lung infections, and problems digesting food. Treatment focuses on airway clearance techniques, enzymes to improve digestion, nutrition, exercise, and antibiotics to treat lung infections. Life expectancy has increased in recent decades but it remains a serious chronic condition.











































![Cystic fibrosis presentation [autosaved] final](https://cdn.slidesharecdn.com/ss_thumbnails/cysticfibrosispresentationautosavedfinal-161019122332-thumbnail.jpg?width=640&height=640&fit=bounds)
















![Cystic fibrosis[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cysticfibrosis1-130307125149-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





























