Downloaded 58 times







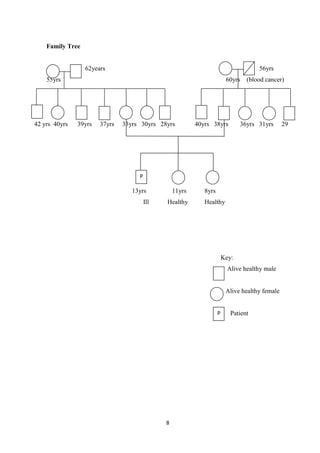






















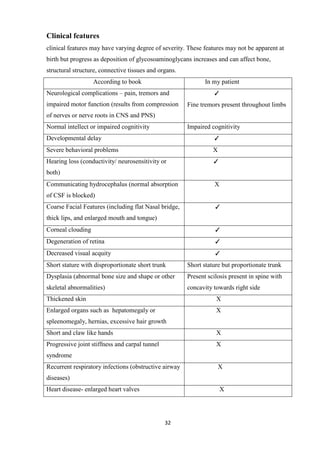


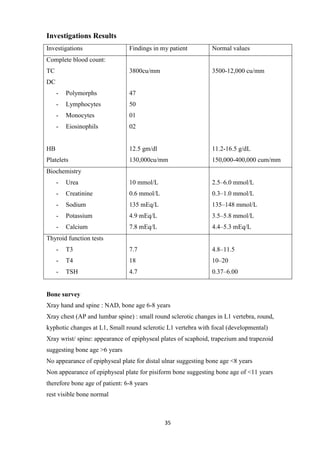
























This case study focuses on developing knowledge of children's growth and health management, particularly for a 13-year-old male patient diagnosed with mucopolysaccharidoses. It outlines the patient's medical history, presenting symptoms, and findings from a comprehensive physical examination, emphasizing his deterioration in health and developmental challenges. The document also describes lysosomal functions and storage disorders, highlighting the impact of enzyme deficiencies on cellular metabolism.



































































