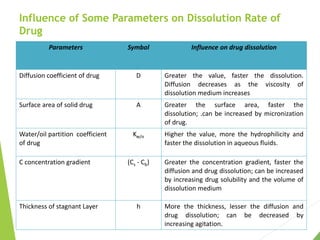
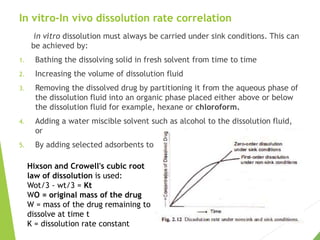
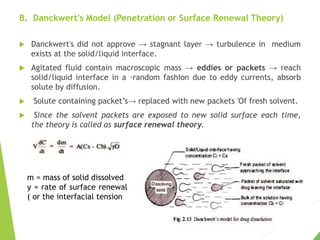
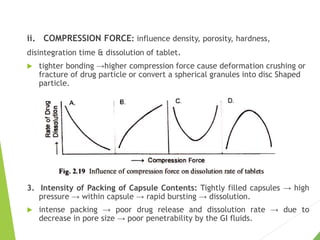
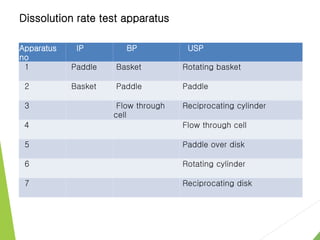
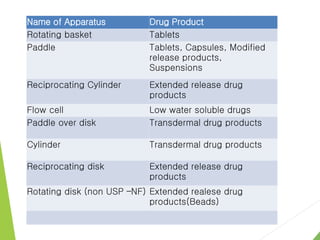
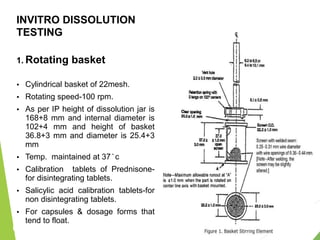
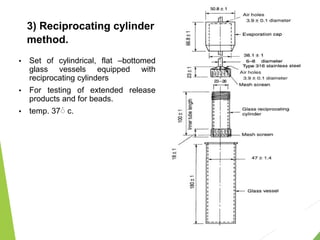
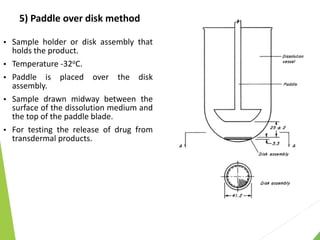
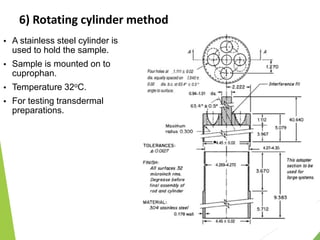
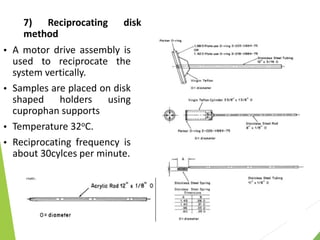
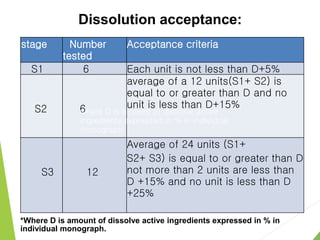
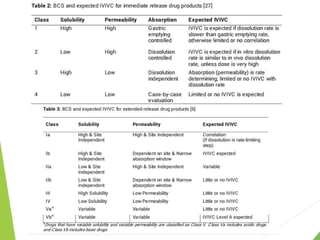
The document discusses the process of dissolution and its importance in drug absorption, detailing the factors influencing dissolution rates such as drug solubility, physicochemical properties, and the manufacturing process. Various theories of drug dissolution, including the diffusion layer model and surface renewal theory, are presented along with the impact of different parameters on dissolution rates. Additionally, it addresses the relationship between drug properties, gastrointestinal pH, and the bioavailability of medications, emphasizing the significance of optimal formulation and in vitro-in vivo correlation for effective drug delivery.