Single Gene Inheritance
AutosomalDominant and Recessive
disorders
Huntigton’s disease, Marfan syndrome, Cystic
fibrosis, Phenylketonuria, Sickle cell disease.
2.
Most humangenetic defects can be categorized
as resulting from either chromosomal, single-
gene Mendelian, single-gene non-Mendelian, or
multifactorial causes.
Disorders associated with single-gene Mendelian
inheritance are typically categorized as
autosomal dominant, autosomal recessive, or
sex-linked.
Modes of Inheritance
3.
In autosomaldominant inheritance, only one copy of a
disease allele is necessary for an individual to be susceptible
to expressing the phenotype.
With each pregnancy, there is a one in two (50%) chance the
offspring will inherit the disease allele.
Unless a new mutation has occurred, all affected individuals
will have at least one parent who is affected by the disease.
Dominantly inherited genetic diseases tend to occur in
every generation of a family.
Dominant mutations can also happen in an individual for
the first time, with no family history of the condition
(spontaneous mutation).
Autosomal Dominant Inheritance
4.
Autosomal dominantinheritance is often called vertical
inheritance because of the transmission from parent to
offspring.
Across a population, the proportion of affected males
should be equal to the proportion of affected females.
Examples of diseases with autosomal dominant
inheritance include Myotonic muscular dystrophy and
Huntington's disease.
Autosomal Dominant Inheritance
5.
In autosomalrecessive inheritance, two copies of a
disease allele are required for an individual to be
susceptible to expressing the phenotype.
Typically, the parents of an affected individual are
not affected but are gene carriers of a mutated
gene.
With each pregnancy of carrier parents there is a
one in four (25%) chance the offspring will inherit
two copies of the disease allele and will therefore
have the phenotype.
Autosomal Recessive Inheritance
6.
There isa one in two (50%) chance the offspring will
inherit one copy of the disease allele and will be a carrier.
There is a one in four (25%) chance the offspring will
inherit no copies of the disease allele and will not express
the phenotype or be a carrier - this individual would not
be at risk for passing the disorder on to his/her offspring.
As with autosomal dominant inheritance, the proportion
of affected males should be equal to the proportion of
affected females in a given population.
Examples of diseases with autosomal recessive
inheritance include sickle cell anemia and cystic fibrosis.
Autosomal Recessive Inheritance
7.
Huntington diseaseis a progressive brain disorder-death of brain
cells - that causes uncontrolled movements, emotional problems, and
loss of thinking ability (cognition).
Adult-onset Huntington disease, the most common form of this
disorder, usually appears in a person's thirties or forties.
Early signs and symptoms can include irritability, depression, small
involuntary movements, poor coordination, and trouble learning new
information or making decisions.
Many people with Huntington disease develop involuntary jerking or
twitching movements known as chorea (dyskinesia).
As the disease progresses, these movements become more
pronounced.
Affected individuals may have trouble walking, speaking, and
swallowing.
Huntington Disease
8.
People withthis disorder also experience changes in
personality and a decline in thinking and reasoning
abilities.
Individuals with the adult-onset form of Huntington
disease usually live about 15 to 20 years after signs and
symptoms begin.
A less common form of Huntington disease known as the
juvenile form begins in childhood or adolescence.
It also involves movement problems and mental and
emotional changes.
9.
Additional signsof the juvenile form include slow
movements, clumsiness, frequent falling, rigidity, slurred
speech, and drooling.
School performance declines as thinking and reasoning
abilities become impaired.
Seizures occur in 30 percent to 50 percent of children
with this condition.
Juvenile Huntington disease tends to progress more
quickly than the adult-onset form
Affected individuals usually live 10 to 15 years after signs
and symptoms appear.
10.
Mutations inthe huntingtin gene (HTT) that is
located on the short arm of chromosome 4, cause
Huntington disease.
The HTT gene provides instructions for making a
protein called huntingtin.
Although the function of this protein is unknown, it
appears to play an important role in nerve cells
(neurons) in the brain.
HTT is expressed in all cells.
Causes
11.
The highestconcentrations are found in the brain
and testes, with moderate amounts in the liver, heart,
and lungs.
It interacts with proteins which are involved in
transcription, cell signaling, and intracellular
transporting.
The huntingtin (HTT) mutation that causes
Huntington disease involves a DNA segment known
as a CAG trinucleotide repeat.
This segment is made up of a series of three DNA
building blocks (cytosine, adenine, and guanine) that
appear multiple times in a row.
12.
Normally, theCAG segment is repeated 10 to 35
times within the gene.
In people with Huntington disease, the CAG
segment is repeated 36 to more than 120 times.
People with 36 to 39 CAG repeats may or may not
develop the signs and symptoms of Huntington
disease, while people with 40 or more repeats
almost always develop the disorder.
13.
An increasein the size of the CAG segment
leads to the production of an abnormally long
version of the huntingtin protein.
The elongated protein is cut into smaller, toxic
fragments that bind together and accumulate
in neurons, disrupting the normal functions of
these cells.
The dysfunction and eventual death of neurons
in certain areas of the brain underlie the signs
and symptoms of Huntington disease.
14.
This conditionis inherited in an autosomal dominant
pattern, which means one copy of the altered gene
in each cell is sufficient to cause the disorder.
An affected person usually inherits the altered gene
from one affected parent.
In rare cases, an individual with Huntington disease
does not have a parent with the disorder.
As the altered HTT gene is passed from one
generation to the next, the size of the CAG
trinucleotide repeat often increases in size.
A larger number of repeats is usually associated
with an earlier onset of signs and symptoms.
This phenomenon is called anticipation.
Inheritance Pattern.
15.
Medical diagnosisof the onset of HD can be made
following the appearance of physical symptoms
specific to the disease.
Genetic testing can be used to confirm a physical
diagnosis if there is no family history of HD.
Even before the onset of symptoms, genetic testing
can confirm if an individual or embryo carries an
expanded copy of the trinucleotide repeat in the
HTT gene that causes the disease.
HD Diagnosis
16.
There isno cure for HD, but there are treatments
available to reduce the severity of some of its
symptoms.
Treatments can relieve some symptoms and in
some improve quality of life.
Full-time care is required in the later stages of the
disease.
Treatment
17.
Current researchdirections include:
◦ determining the exact mechanism of the
disease.
◦ improving animal models to aid with research.
◦ testing of medications to treat symptoms or
slow the progression of the disease.
◦ studying procedures such as stem cell therapy
with the goal of repairing damage caused by
the disease.
18.
Marfan syndrome(MFS) is a genetic disorder of the
connective tissue.
It is named after Antoine Marfan, a French pediatrician who
first described the condition in 1896.
MFS is an autosomal dominant disorder
About 1 in 5,000 to 10,000 individuals have Marfan
syndrome.
It occurs equally in males and females, rates are similar
between races and in different regions of the world.
About 75% of the time, the condition is inherited from a
parent, while 25% of the time it is a new mutation.
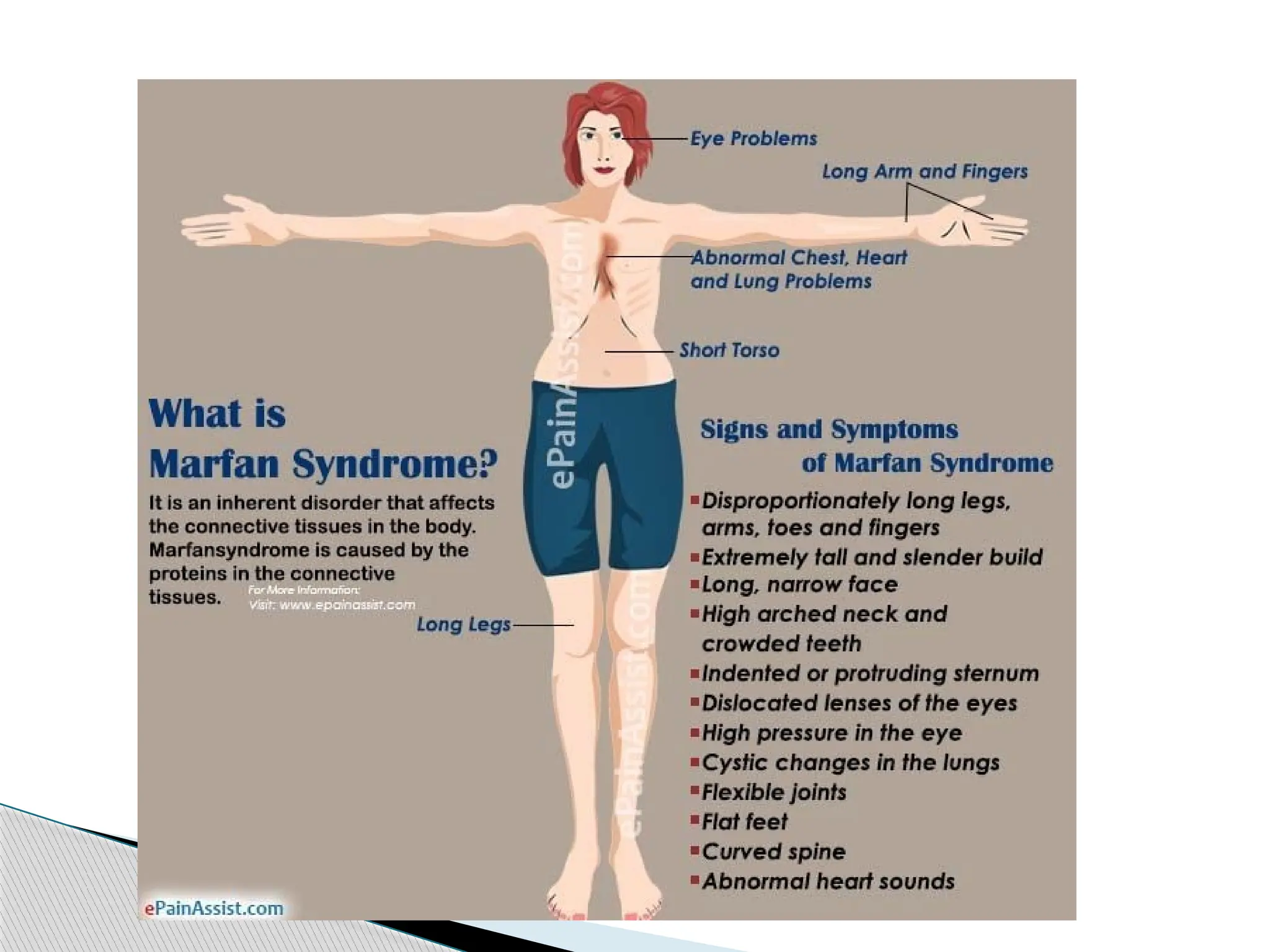
Marfan Syndrome (MFS)
19.
More than30 different signs and symptoms are
variably associated with Marfan syndrome.
The most prominent of these affect the skeletal,
cardiovascular, and ocular systems, but all fibrous
connective tissue throughout the body can be
affected.
Signs and Symptoms
20.
People withMarfan tend to be tall and thin, with long arms,
legs, fingers and toes.
They also typically have flexible joints and scoliosis.
The most serious complications involve the heart and aorta,
with an increased risk of mitral valve prolapse and aortic
aneurysm.
Other commonly affected areas include the lungs, eyes, bones
and the covering of the spinal cord.
It involves a mutation to the gene that makes fibrillin, which
results in abnormal connective tissue.
A diagnosis is based on family history and a combination of
major and minor indicators of the disorder.
Diagnostic criteria of MFS - Ghent nosology - were agreed upon
internationally in 1996 and then revised in 2010.
22.
There isno known cure for Marfan syndrome.
Many people have a normal life expectancy with
proper treatment.
Management often includes the use of beta
blockers.
Surgery may be required to repair the aorta or
replace a heart valve.
It is recommended that energetic exercise be
avoided.
Regular checkups are recommended to monitor
the health of the heart valves and the aorta.
Management.
23.
Cystic fibrosisis an inherited disease characterized by the buildup
of thick, sticky mucus that can damage many of the body's organs.
The disease occurs in 1 in 2,500 to 3,500 white newborns. Cystic
fibrosis is less common in other ethnic groups, affecting about 1 in
17,000 African Americans and 1 in 31,000 Asian Americans.
The most common signs and symptoms include progressive
damage to the respiratory system and chronic digestive system
problems.
The features of the disorder and their severity varies among
affected individuals.
In people with cystic fibrosis, the body produces mucus that is
abnormally thick and sticky.
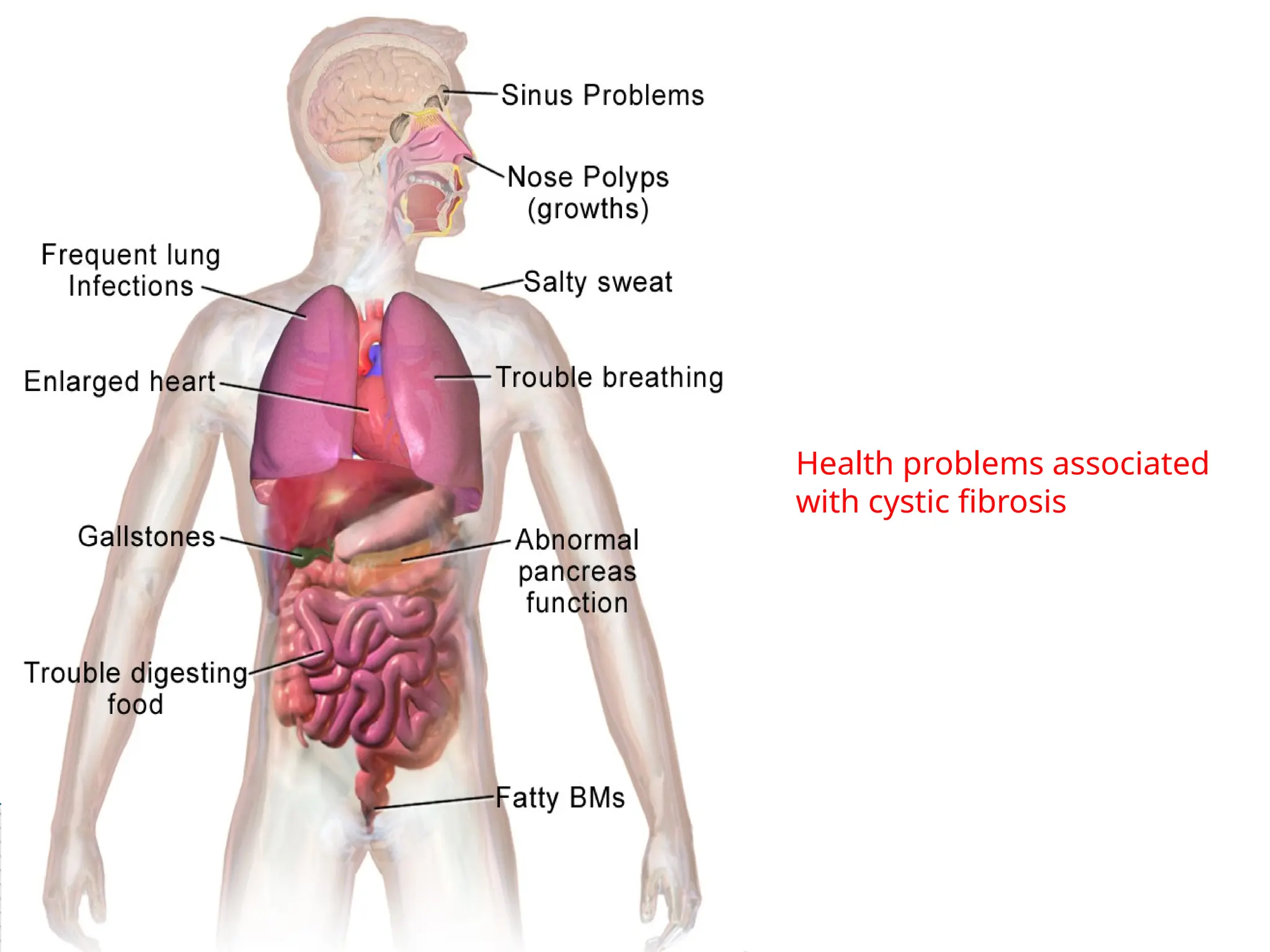
Cystic Fibrosis
24.
This abnormalmucus can clog the airways,
leading to severe problems with breathing and
bacterial infections in the lungs.
These infections cause chronic coughing,
wheezing, and inflammation.
Over time, mucus buildup and infections result
in permanent lung damage, including the
formation of scar tissue (fibrosis) and cysts in
the lungs.
25.
In peoplewith cystic fibrosis, mucus often
damages the pancreas, impairing its ability to
produce insulin and digestive enzymes.
Problems with digestion can lead to diarrhea,
malnutrition, poor growth, and weight loss.
In adolescence or adulthood, a shortage of
insulin can cause a form of diabetes known as
cystic fibrosis-related diabetes mellitus
(CFRDM).
26.
Cystic fibrosisused to be considered a fatal
disease of childhood.
Adults with cystic fibrosis experience health
problems affecting the respiratory, digestive, and
reproductive systems.
Most men with cystic fibrosis have congenital
bilateral absence of the vas deferens (CBAVD), a
condition in which the tubes that carry sperm are
blocked by mucus and do not develop properly,
causing infertility unless the fertility treatment.
Women with cystic fibrosis may experience
complications in pregnancy.
Mutations inthe transmembrane conductance regulator
(CFTR gene) on chromosome 7 cause cystic fibrosis.
The CFTR gene provides instructions for making a
channel that transports negatively charged particles
called chloride ions into and out of cells.
Chloride is a component of sodium chloride, a common
salt found in sweat.
Chloride also has important functions in cells; for
example, the flow of chloride ions helps control the
movement of water in tissues, which is necessary for the
production of thin, freely flowing mucus.
Causes
29.
Mutations inthe CFTR gene disrupt the function
of the chloride channels, preventing them from
regulating the flow of chloride ions and water
across cell membranes.
As a result, cells that line the passageways of the
lungs, pancreas, and other organs produce
mucus that is unusually thick and sticky.
This mucus clogs the airways and various ducts,
causing the characteristic signs and symptoms
of cystic fibrosis.
30.
Other geneticand environmental factors likely
influence the severity of the condition.
For example, mutations in genes other than
CFTR might help explain why some people with
cystic fibrosis are more severely affected than
others.
Most of these genetic changes have not been
identified, however.
31.
This conditionis inherited in an autosomal recessive
pattern, which means both copies of the gene in each cell
have mutations.
The parents of an individual with an autosomal recessive
condition each carry one copy of the mutated gene, but
they typically do not show signs and symptoms of the
condition.
If two carriers have a child, there is a:
◦ 25 percent, or 1 in 4, chance the child will have CF
◦ 50 percent, or 1 in 2, chance the child will be a carrier but will not
have CF
◦ 25 percent, or 1 in 4, chance the child will not be a carrier and will
not have CF
Inheritance Pattern
32.
Cystic fibrosismay be diagnosed by many different methods, including
newborn screening, sweat testing, and genetic testing.
There is currently no cure for CF.
Treatment can manage the symptoms of the disease, however, and
improve quality of life.
It is crucial for people with CF to get rid of mucus from their lungs to
allow clear breathing and minimize lung infections.
Inhaled medication is effective at reaching the airways and commonly
used.
Antibiotics are an important part of regular care.
Gene therapy has been explored as a potential cure for CF. Results from
clinical trials have shown limited success as of 2016, and using gene
therapy as routine therapy is not suggested.
Diagnosis and Management
33.
Phenylketonuria (PKU)is an inherited disorder with
decreased metabolism of the amino acid phenylalanine,
resulting in increased levels of this amino acid in the
blood.
This condition is inherited in an autosomal recessive
pattern, which means both copies of the gene in each cell
have mutations.
The parents of an individual with an autosomal recessive
condition each carry one copy of the mutated gene, but
they typically do not show signs and symptoms of the
condition.
Phenilketonuria.
34.
Phenylalanine isa building block of proteins that
is obtained through the diet.
When Phe cannot be metabolized by the body, a
typical diet that would be healthy for people
without PKU causes abnormally high levels of Phe
to accumulate in the blood, which is toxic to the
brain.
If PKU is not treated, phenylalanine can build up
to harmful levels in the body, causing intellectual
disability and other serious health problems.
The signs and symptoms of PKU vary from mild to
severe.
35.
The mostsevere form of this disorder is known as
classic PKU.
Infants with classic PKU appear normal until they
are a few months old.
Without treatment, these children develop
permanent intellectual disability.
Seizures, delayed development, behavioral
problems, and psychiatric disorders are also
common.
36.
Untreated individualsmay have a musty or mouse-
like odor as a side effect of excess phenylalanine in
the body.
Children with classic PKU tend to have lighter skin
and hair than unaffected family members and are
also likely to have skin disorders such as eczema.
37.
Less severeforms of this condition, sometimes
called variant PKU and non-PKU
hyperphenylalaninemia, have a smaller risk of
brain damage.
People with very mild cases may not require
treatment with a low-phenylalanine diet.
38.
Babies bornto mothers who have PKU and
uncontrolled phenylalanine levels (women who no
longer follow a low-phenylalanine diet) have a
significant risk of intellectual disability because
they are exposed to very high levels of
phenylalanine before birth.
These infants may also have a low birth weight and
grow more slowly than other children.
Mutations inthe PAH gene cause phenylketonuria.
The PAH gene provides instructions for making an enzyme
called phenylalanine hydroxylase.
This enzyme converts the amino acid phenylalanine to other
important amino acid tyrosine.
If gene mutations reduce the activity of phenylalanine
hydroxylase, phenylalanine from the diet is not processed
effectively.
Phenylalanine accumulates and is converted into
phenylpyruvate (also known as phenylketone), which can be
detected in the urine.
As a result, this amino acid can build up to toxic levels in the
blood and other tissues.
Because nerve cells in the brain are particularly sensitive to
phenylalanine levels, excessive amounts of this substance can
cause brain damage.
Causes
41.
Classic PKU,the most severe form of the disorder, occurs
when phenylalanine hydroxylase activity is severely
reduced or absent.
People with untreated classic PKU have levels of
phenylalanine high enough to cause severe brain damage
and other serious health problems.
Mutations in the PAH gene that allow the enzyme to retain
some activity result in milder versions of this condition,
such as variant PKU or non-PKU hyperphenylalaninemia.
Changes in other genes may influence the severity of PKU,
but little is known about these additional genetic factors.
42.
PKU iscommonly included in the newborn screening
panel of many countries, with varied detection
techniques.
Most babies in developed countries are screened for
PKU soon after birth.
PKU is not curable.
However, if PKU is diagnosed early enough, an affected
newborn can grow up with normal brain development by
managing and controlling phenylalanine ("Phe") levels
through diet, or a combination of diet and medication.
Diagnosis and Management
43.
People whofollow the prescribed dietary
treatment from birth may have no symptoms.
Their PKU would be detectable only by a blood
test.
People must adhere to a special diet low in Phe
for optimal brain development.
Since Phe is necessary for the synthesis of
many proteins, it is required for appropriate
growth, but levels must be strictly controlled.
44.
The dietrequires restricting or eliminating
foods high in Phe, such as soybeans, egg
whites, shrimp, chicken breast, spirulina,
watercress, fish, nuts, crayfish, lobster, tuna,
turkey, legumes, and lowfat cottage cheese.
Starchy foods, such as potatoes and corn are
generally acceptable in controlled amounts, but
the quantity of Phe consumed from these
foods must be monitored.
45.
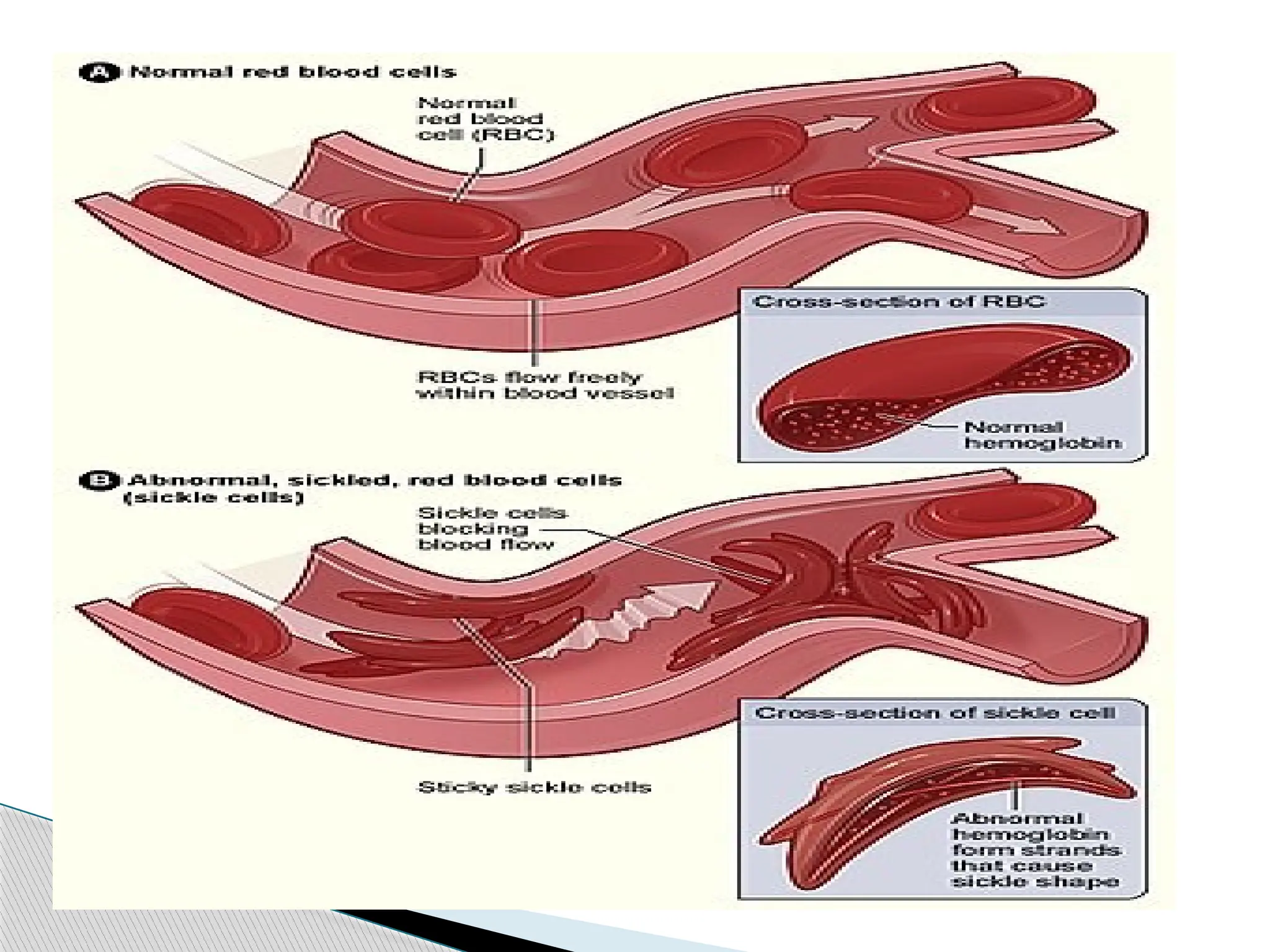
Sickle celldisease is a group of disorders that affects
hemoglobin, the molecule in red blood cells that delivers
oxygen to cells throughout the body.
This condition is inherited in an autosomal recessive pattern.
People with this disorder have atypical hemoglobin
molecules called hemoglobin S, which can distort red blood
cells into a sickle, or crescent, shape.
Signs and symptoms of sickle cell disease usually begin in
early childhood.
Characteristic features of this disorder include a low number
of red blood cells (anemia), repeated infections, and periodic
episodes of pain.
Sickle Cell Disease
46.
The severityof symptoms varies from person to
person.
The signs and symptoms of sickle cell disease are
caused by the sickling of red blood cells.
When red blood cells sickle, they break down
prematurely, which can lead to anemia
Anemia can cause shortness of breath, fatigue, and
delayed growth and development in children.
The rapid breakdown of red blood cells may also
cause yellowing of the eyes and skin, which are
signs of jaundice.
47.
Painful episodescan occur when sickled red blood cells,
which are stiff and inflexible, get stuck in small blood
vessels.
These episodes deprive tissues and organs of oxygen-
rich blood and can lead to organ damage, especially in
the lungs, kidneys, spleen, and brain.
A particularly serious complication of sickle cell disease
is high blood pressure in the blood vessels that supply
the lungs (pulmonary hypertension).
Pulmonary hypertension occurs in about one-third of
adults with sickle cell disease and can lead to heart
failure.
48.
Sickle celldisease affects millions of people worldwide.
It is most common among people whose ancestors
come from Africa; Mediterranean countries such as
Greece, Turkey, and Italy; the Arabian Peninsula; India;
and Spanish-speaking regions in South America, Central
America, and parts of the Caribbean.
The disease is estimated to occur in 1 in 500 African
Americans and 1 in 1,000 to 1,400 Hispanic Americans.
Frequency
50.
Mutations inthe HBB gene (chromosome 11)
cause sickle cell disease.
Hemoglobin consists of four protein subunits,
typically, two subunits called alpha-globin and two
subunits called beta-globin.
The HBB gene provides instructions for making
beta-globin.
Various versions of beta-globin result from
different mutations in the HBB gene.
Causes
51.
One particularHBB gene mutation produces an
abnormal version of beta-globin known as
hemoglobin S (HbS).
Other mutations in the HBB gene lead to
additional abnormal versions of beta-globin such
as hemoglobin C (HbC) and hemoglobin E (HbE).
HBB gene mutations can also result in an
unusually low level of beta-globin.
This abnormality is called beta thalassemia.
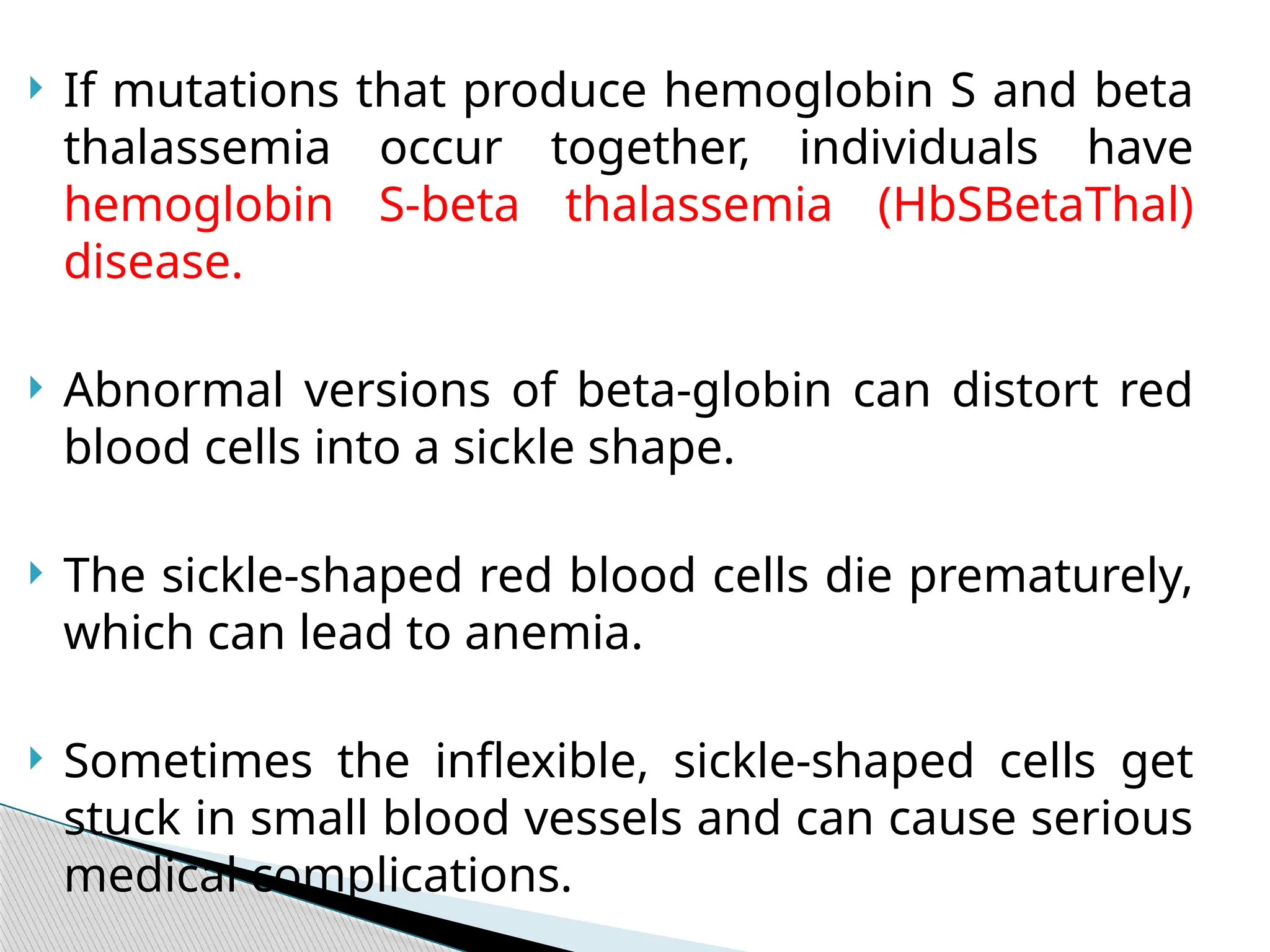
52.
If mutationsthat produce hemoglobin S and beta
thalassemia occur together, individuals have
hemoglobin S-beta thalassemia (HbSBetaThal)
disease.
Abnormal versions of beta-globin can distort red
blood cells into a sickle shape.
The sickle-shaped red blood cells die prematurely,
which can lead to anemia.
Sometimes the inflexible, sickle-shaped cells get
stuck in small blood vessels and can cause serious
medical complications.
53.
In HbS,the complete blood count reveals low
hemoglobin levels with a high reticulocyte count.
Abnormal hemoglobin forms can be detected on
hemoglobin electrophoresis.
Sickle cell hemoglobin (HgbS) and hemoglobin C
with sickling (HgbSC)—the two most common
forms—can be identified from there.
Diagnosis
54.
Albinism isa genetic disorder characterized in
humans by the complete or partial absence of
pigment in the skin, hair and eyes.
Albinism is associated with a number of vision
defects.
Lack of skin pigmentation makes for more
susceptibility to sunburn and skin cancers.
Albinism results from inheritance of recessive
gene alleles and is known to affect all vertebrates,
including humans.
Albinism
55.
It isdue to absence or defect of tyrosinase, a
copper-containing enzyme involved in the
production of melanin.
Albinism is considered to be a hereditary
condition characterised by the absence of
melanin in particular, in the eyes, skin, hair or
feathers.
While an organism with complete absence of
melanin is called an albino, an organism with
only a diminished amount of melanin is
described as albinoid.
56.
There aretwo principal types of albinism:
oculocutaneous, affecting the eyes, skin and hair,
and ocular affecting the eyes only.
Because individuals with albinism have skin that
entirely lacks the dark pigment melanin, which
helps protect the skin from the sun's ultraviolet
radiation, their skin can burn more easily from
overexposure.
Those with albinism are generally as healthy as the
rest of the population, with growth and
development occurring as normal, and albinism by
itself does not cause mortality
Although the lack of pigment blocking ultraviolet
radiation increases the risk of melanomas (skin
cancers) and other problems.
58.
Oculocutaneous albinismis generally the result of
the biological inheritance of genetically recessive
alleles (genes) passed from both parents of an
individual for example OCA1 and OCA2.
Some rare forms are inherited from only one parent.
There are other genetic mutations which are proven
to be associated with albinism.
All alterations, however, lead to changes in melanin
production in the body
Some of these are associated with increased risk of
skin cancer.
Causes
59.
The chanceof offspring with albinism resulting from
the pairing of an organism with albinism and one
without albinism is low.
Albinism usually occurs with equal frequency in both
sexes.
An exception to this is ocular albinism, which it is
passed on to offspring through X-linked inheritance.
Thus, ocular albinism occurs more frequently in males
as they have a single X and Y chromosome, unlike
females, whose genetics are characterized by two X
chromosomes.
60.
People withalbinism will have the following symptoms:
◦ an absence of color in the hair, skin, or eyes
◦ lighter than normal coloring of the hair, skin, or eyes
◦ patches of skin that have an absence of color
Albinism occurs with vision problems, which may
include:
◦ strabismus (crossed eyes)
◦ photophobia (sensitivity to light)
◦ nystagmus (involuntary rapid eye movements)
◦ impaired vision or blindness
◦ astigmatism
Symptoms
61.
Since thereis no cure for albinism, it is
managed through lifestyle adjustments.
People with albinism need to take care not to
get sunburnt and should have regular healthy
skin checks by a dermatologist.
Most forms of albinism don’t affect life span.
Management







































































































![[BROCHURE] Italy Tour Project | @SlideON](https://cdn.slidesharecdn.com/ss_thumbnails/brochure8-251215152319-2805af68-thumbnail.jpg?width=640&height=640&fit=bounds)