Downloaded 11 times



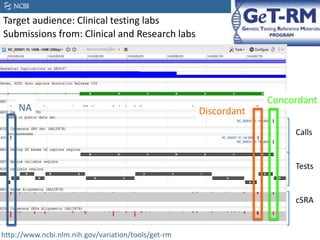

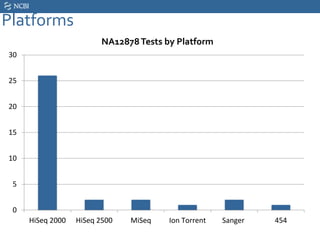

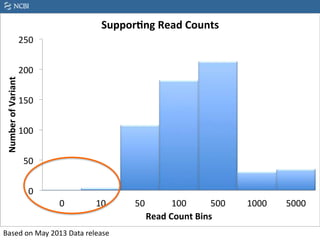
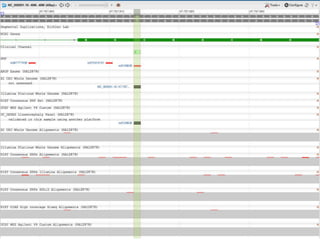
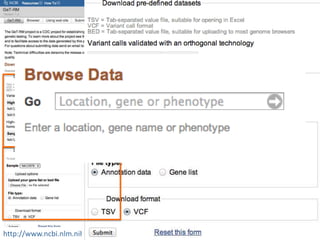
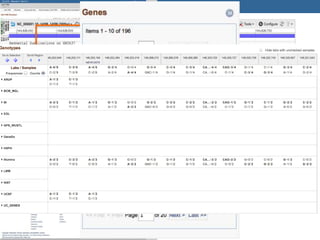
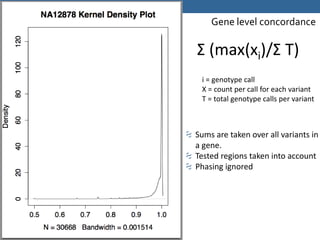


This document summarizes the GeT-RM Project and Browser, which collects and analyzes genetic variant data submitted by various clinical and research laboratories. It describes the project team members and submitting labs. It provides information on the types and platforms of tests conducted, validation methods used by labs, read count distributions, and gene-level concordance calculations. It also outlines future analyses, improvements to the browser, and inclusion of paralogous sequence variant data.



































































