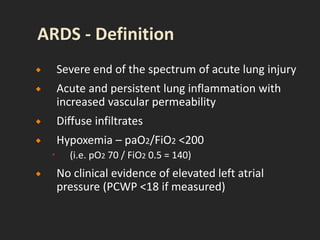
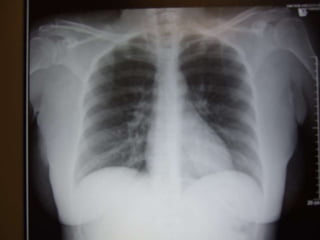
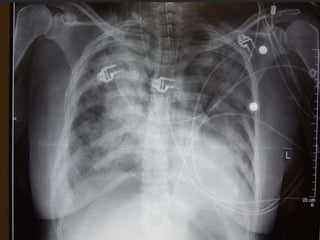
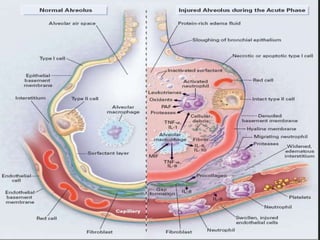
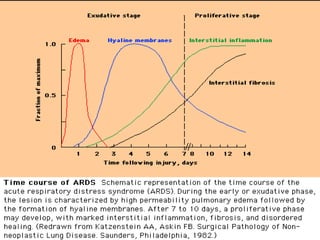
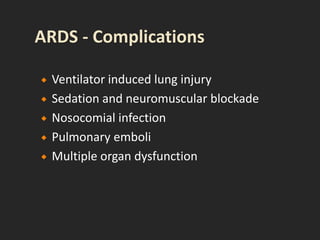
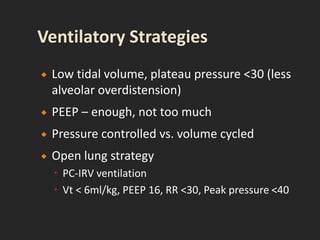
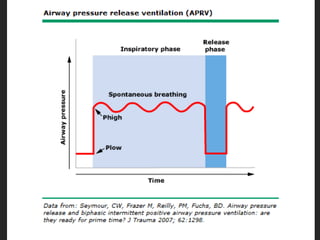
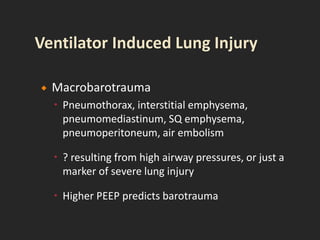
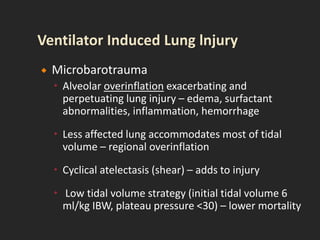
This document discusses respiratory failure and acute respiratory distress syndrome (ARDS). It provides a historical overview of developments in understanding and treating respiratory failure. It describes the types and causes of respiratory failure and defines ARDS. Pathophysiology, risk factors, stages, and management strategies for ARDS are outlined. Ventilator strategies aim to provide oxygenation while avoiding additional lung injury. Low tidal volume ventilation has reduced ARDS mortality.
















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)














![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)


