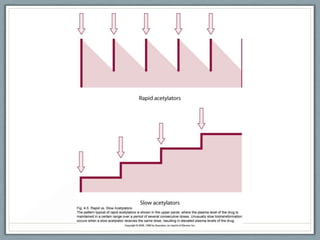
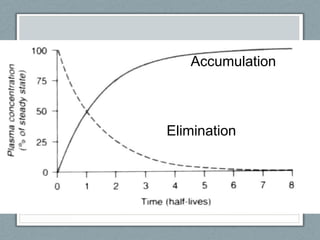
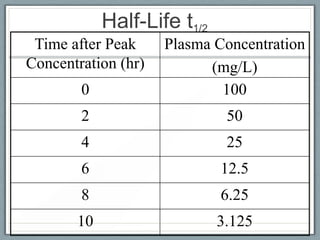
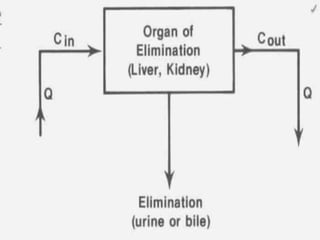
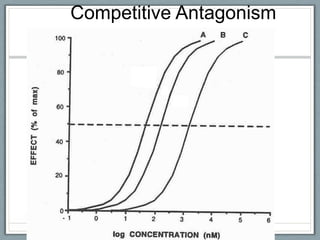
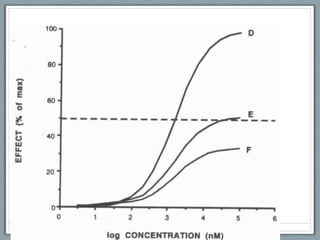
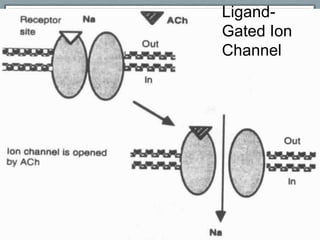
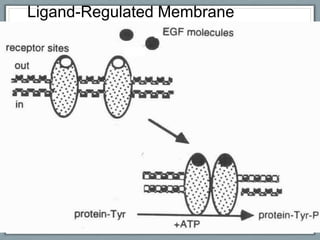
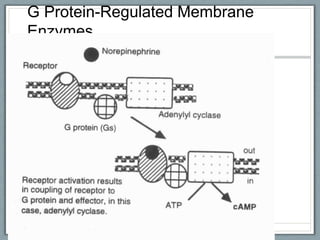
Pharmacodynamics is the study of how drugs act on biological systems and their mechanisms of action. Drugs can interact with receptors to mimic or block physiological messengers. Agonists activate receptors while antagonists reduce or prevent agonist effects. Receptors are often proteins that drugs bind to through covalent, ionic, or hydrogen bonds. Signaling mechanisms involve ligand-gated ion channels, G-protein coupled receptors, and second messengers such as cAMP or calcium ions. Understanding these interactions is important for elucidating drug actions at the cellular level.