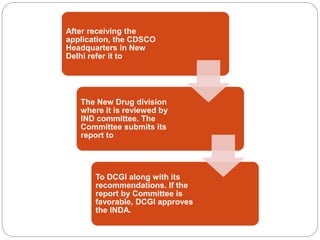
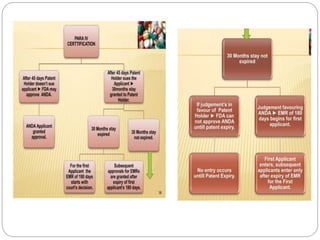
The document discusses Investigational New Drug (IND) applications and Abbreviated New Drug Applications (ANDAs). It defines an IND as a new drug or biological used in clinical investigation. The sponsor submits an IND application to the FDA or DCGI to begin clinical trials in humans. Clinical trials can start 30 days after IND submission if not objected to by the FDA. ANDAs allow for generic drug approval without full clinical trials, as long as they are equivalent to an existing brand drug in dosage, strength, quality and performance. The process for IND and ANDA approval in both the US and India is described.