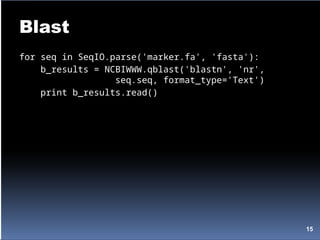
Biopython is a set of tools for computational molecular biology designed to facilitate bioinformatics programming in Python through reusable modules. It allows for the manipulation of DNA and protein sequences, access to public databases, and various genetic analyses. The document includes examples of working with biological data formats, including parsing FASTA files and running BLAST queries.





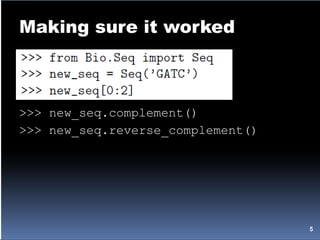

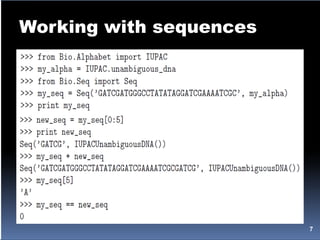
![Working with sequences
>>> protein_seq = Seq('EVRNAK', IUPAC.protein)
>>> dna_seq = Seq('ACGT', IUPAC.unambiguous_dna)
>>> protein_seq + dna_seq
>>> my_seq.tostring()
>>> my_seq[5] = 'G
>>> mutable_seq = my_seq.tomutable()
>>> print mutable_seq
>>> mutable_seq[5] = 'T'
>>> print mutable_seq
>>> mutable_seq.remove('T')
>>> print mutable_seq
>>> mutable_seq.reverse()
>>> print mutable_seq
8](https://image.slidesharecdn.com/10518261-240811020935-4ac6614c/85/10518261_biopython_python_slides_notes-ppt-8-320.jpg)
![Parsing biological file formats
>gi|6273290|gb|AF191664.1|AF191664 Opuntia clavata rpl16 gene; chloroplast
gene for...
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAGAAAGAAAAAAATGAA
TCTAAATGATATAGGATTCCACTATGTAAGGTCTTTGAATCATATCATAAAAGACAATGTAAT
AAA...
import string
from Bio.ParserSupport import AbstractConsumer
class SpeciesExtractor(AbstractConsumer):
def __init__(self):
self.species_list = []
def title(self, title_info):
title_atoms = string.split(title_info)
new_species = title_atoms[1]
if new_species not in self.species_list:
self.species_list.append(new_species)
9](https://image.slidesharecdn.com/10518261-240811020935-4ac6614c/85/10518261_biopython_python_slides_notes-ppt-9-320.jpg)
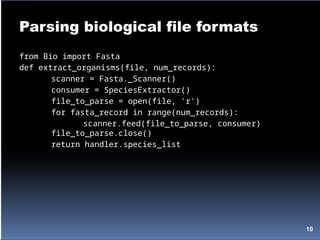
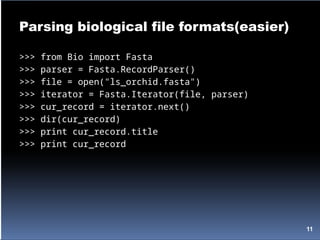
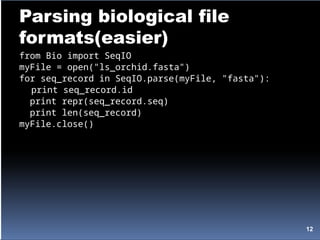
![FASTA files as Dictionaries
import string
def get_accession_num(fasta_record):
title_atoms = string.split(fasta_record.title)
# all of the accession number information is stuck
in the first element
# and separated by '|'s
accession_atoms = string.split(title_atoms[0], '|')
# the accession number is the 4th element
gb_name = accession_atoms[3]
# strip the version info before returning
return gb_name[:-2]
13](https://image.slidesharecdn.com/10518261-240811020935-4ac6614c/85/10518261_biopython_python_slides_notes-ppt-13-320.jpg)