



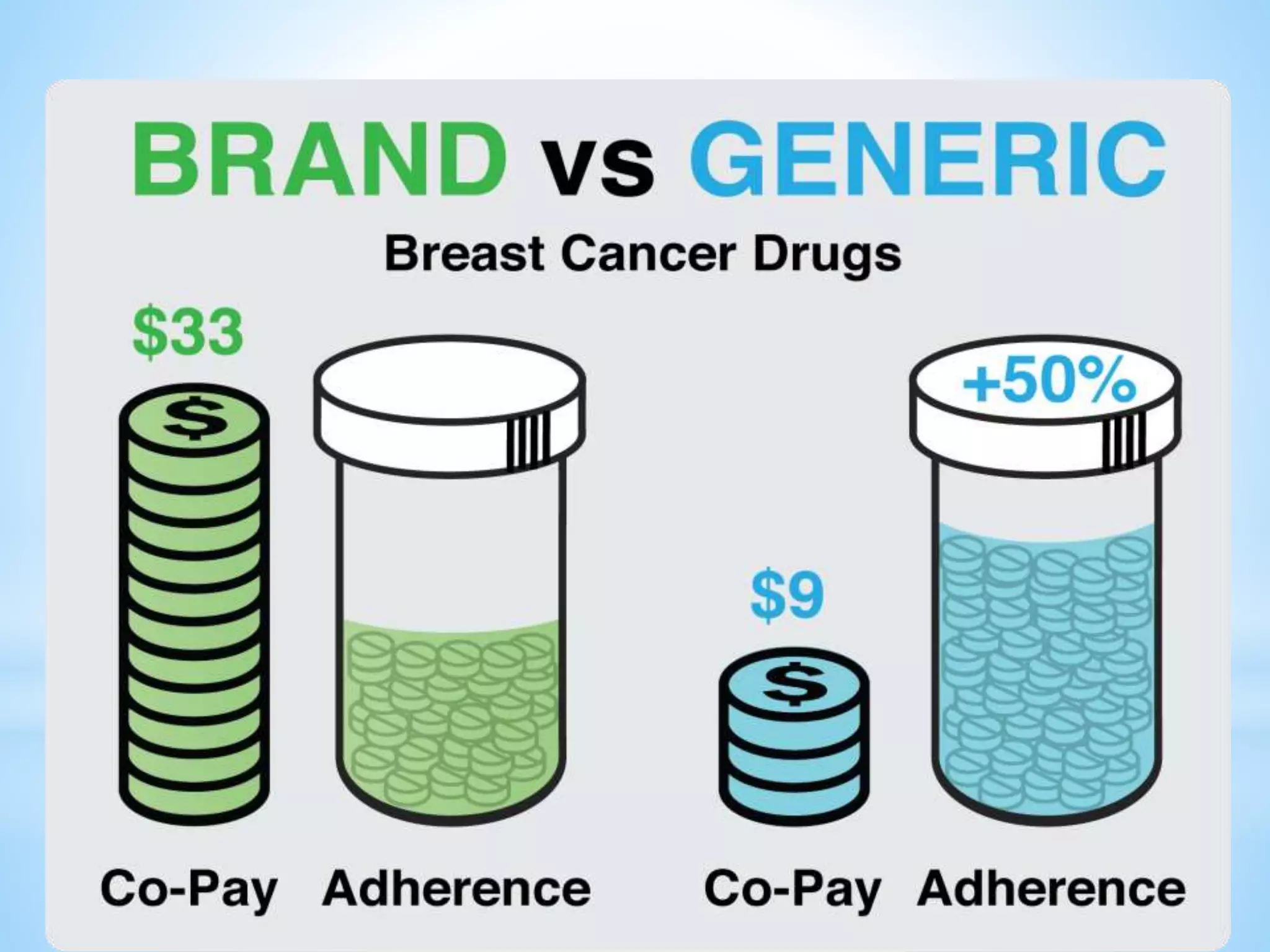



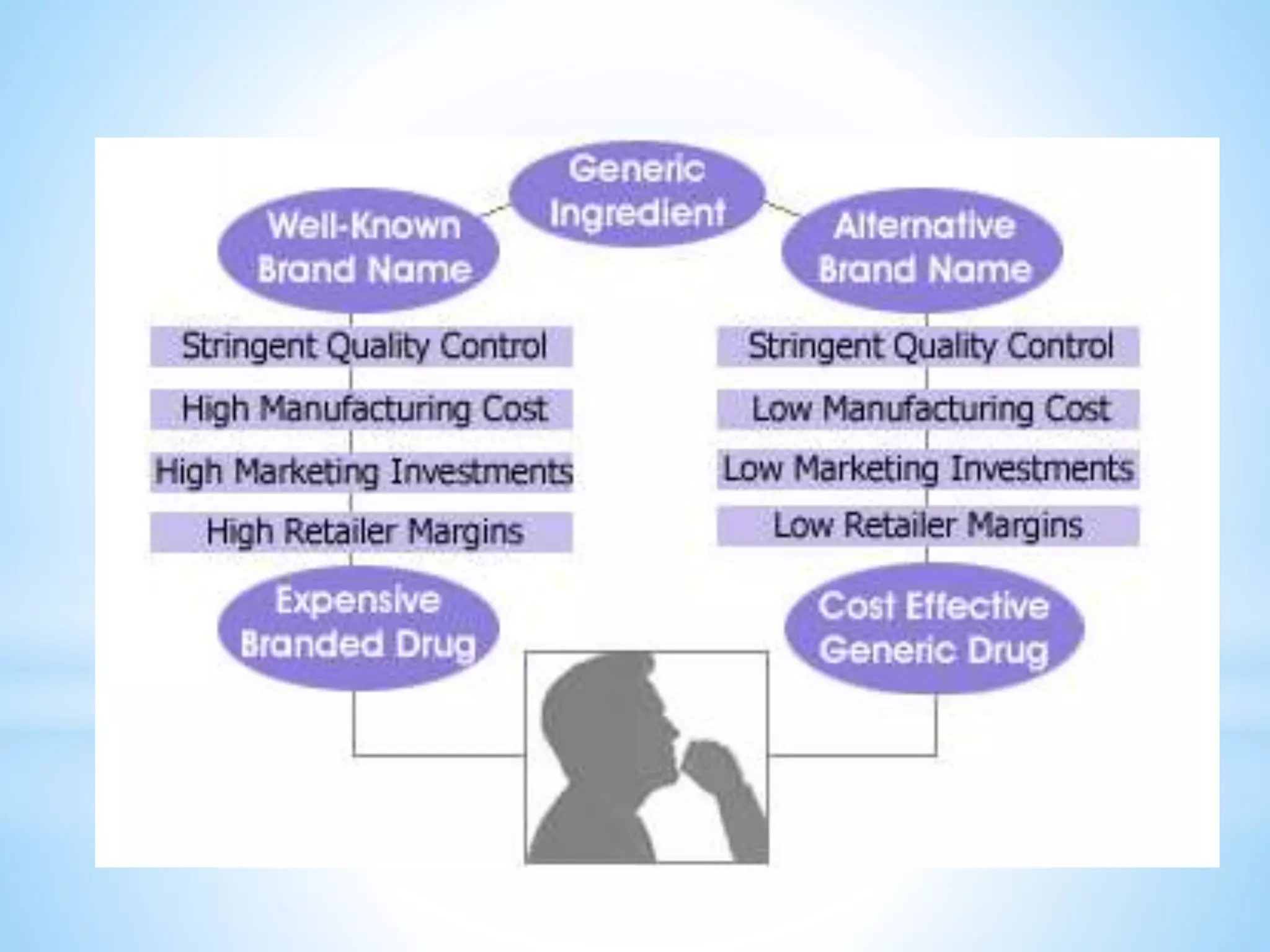





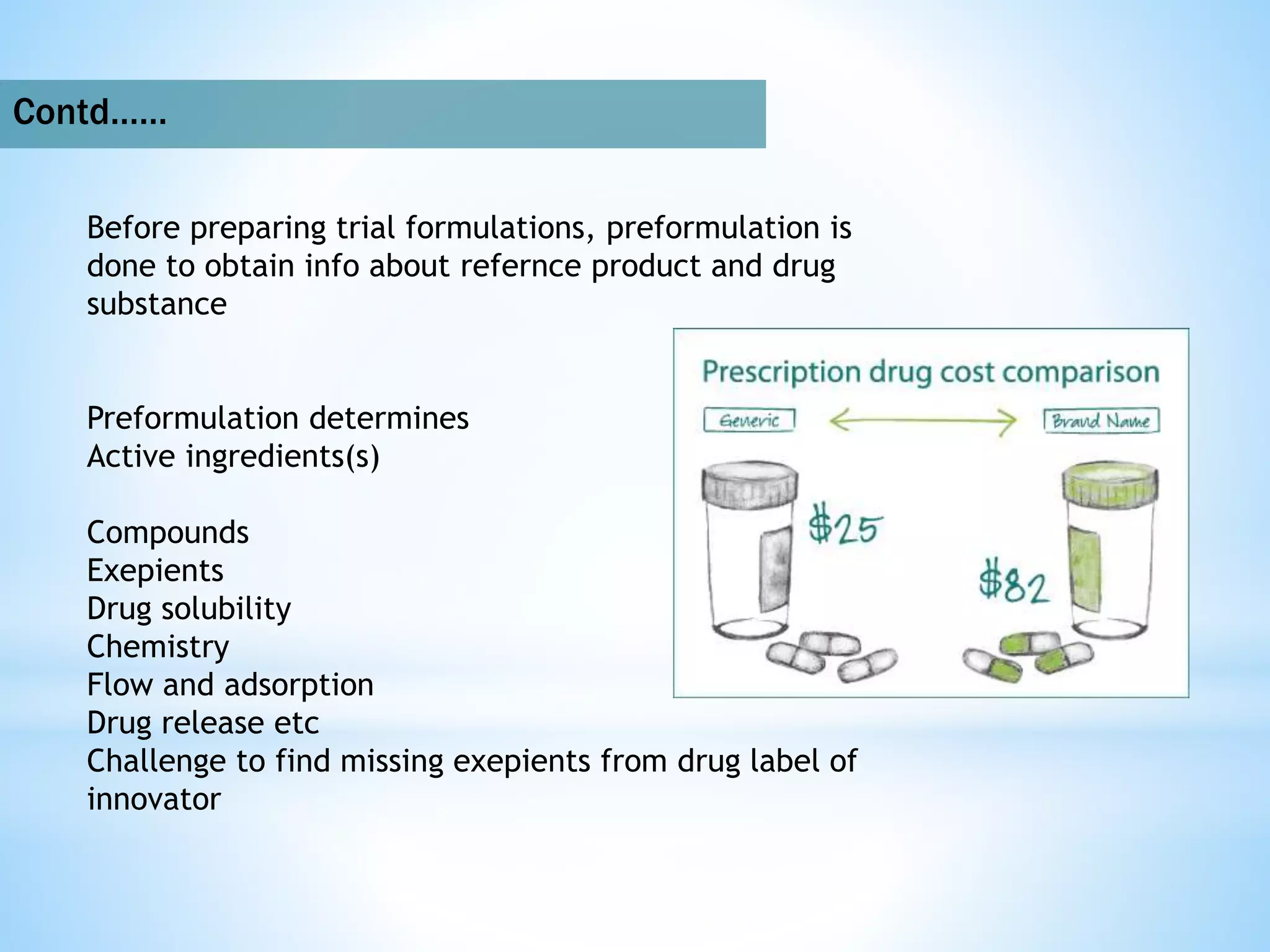


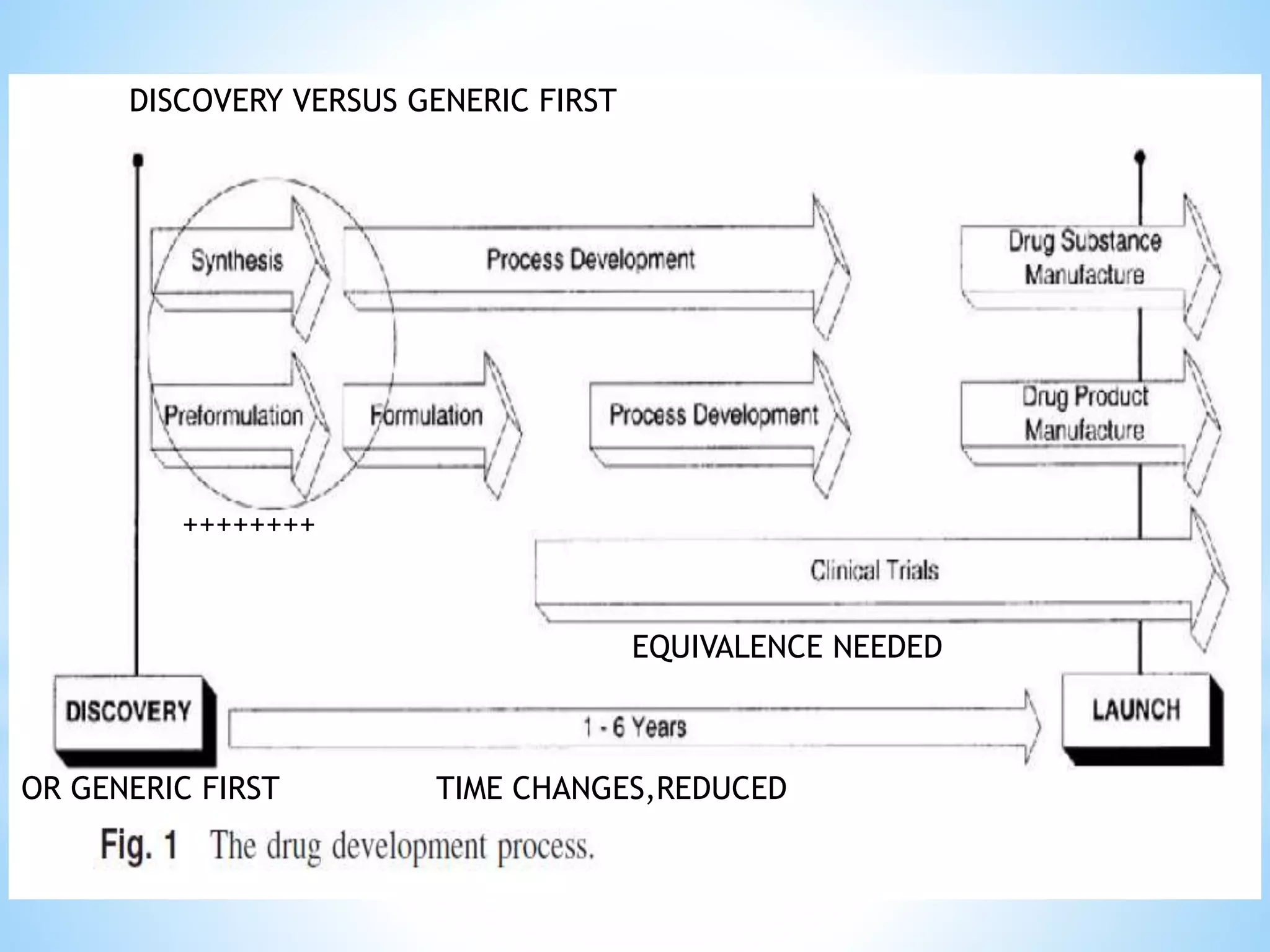
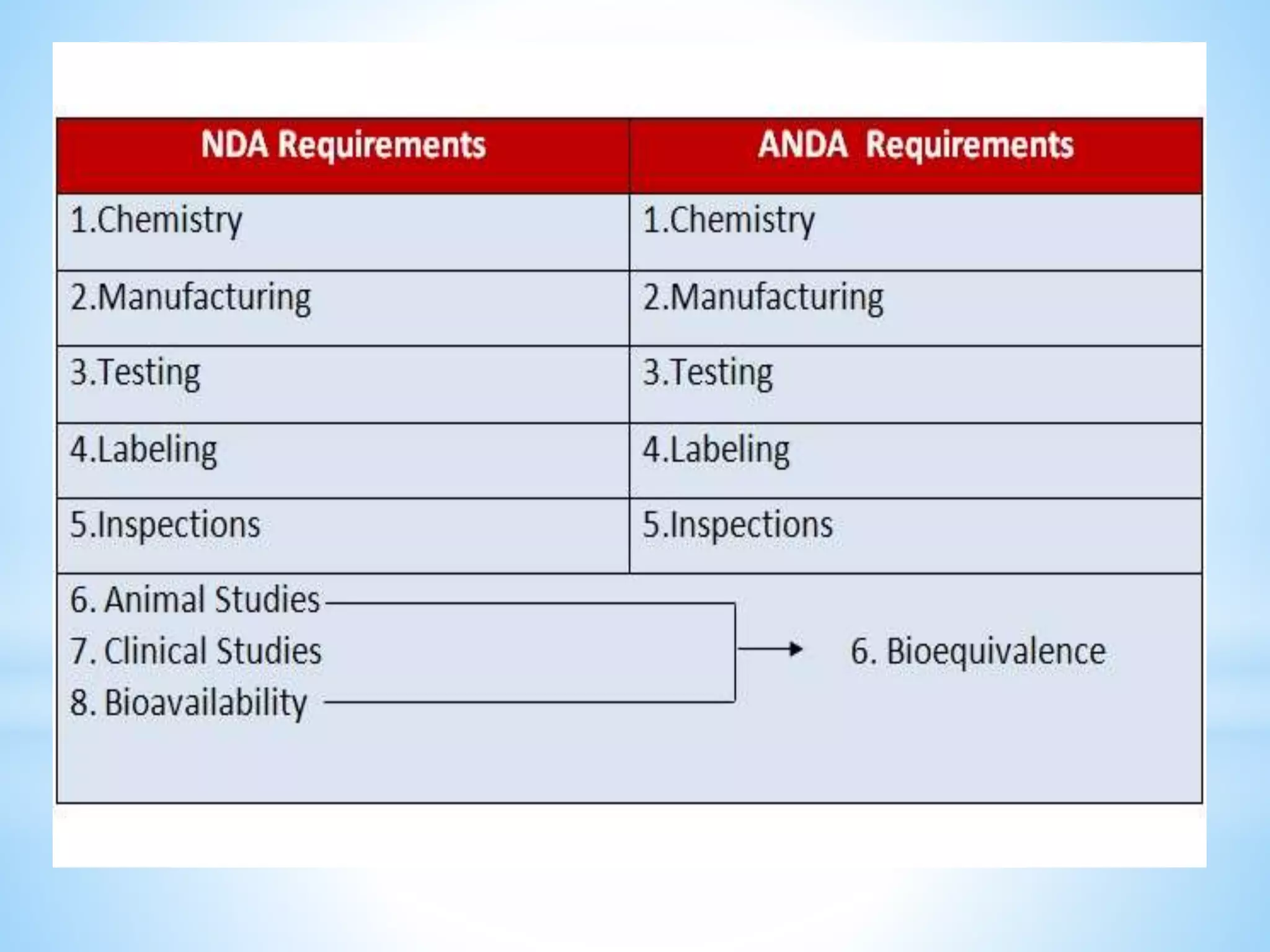
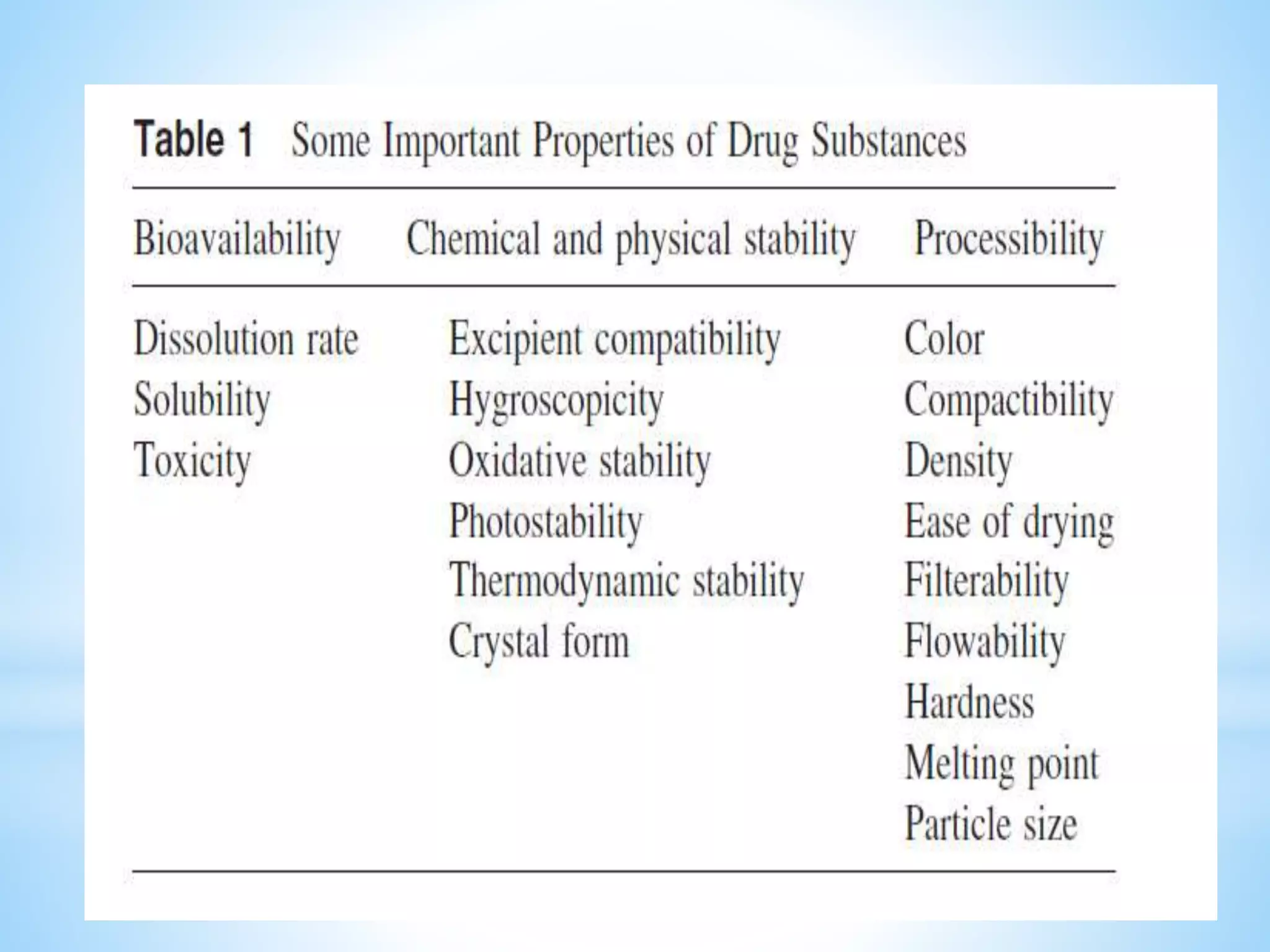





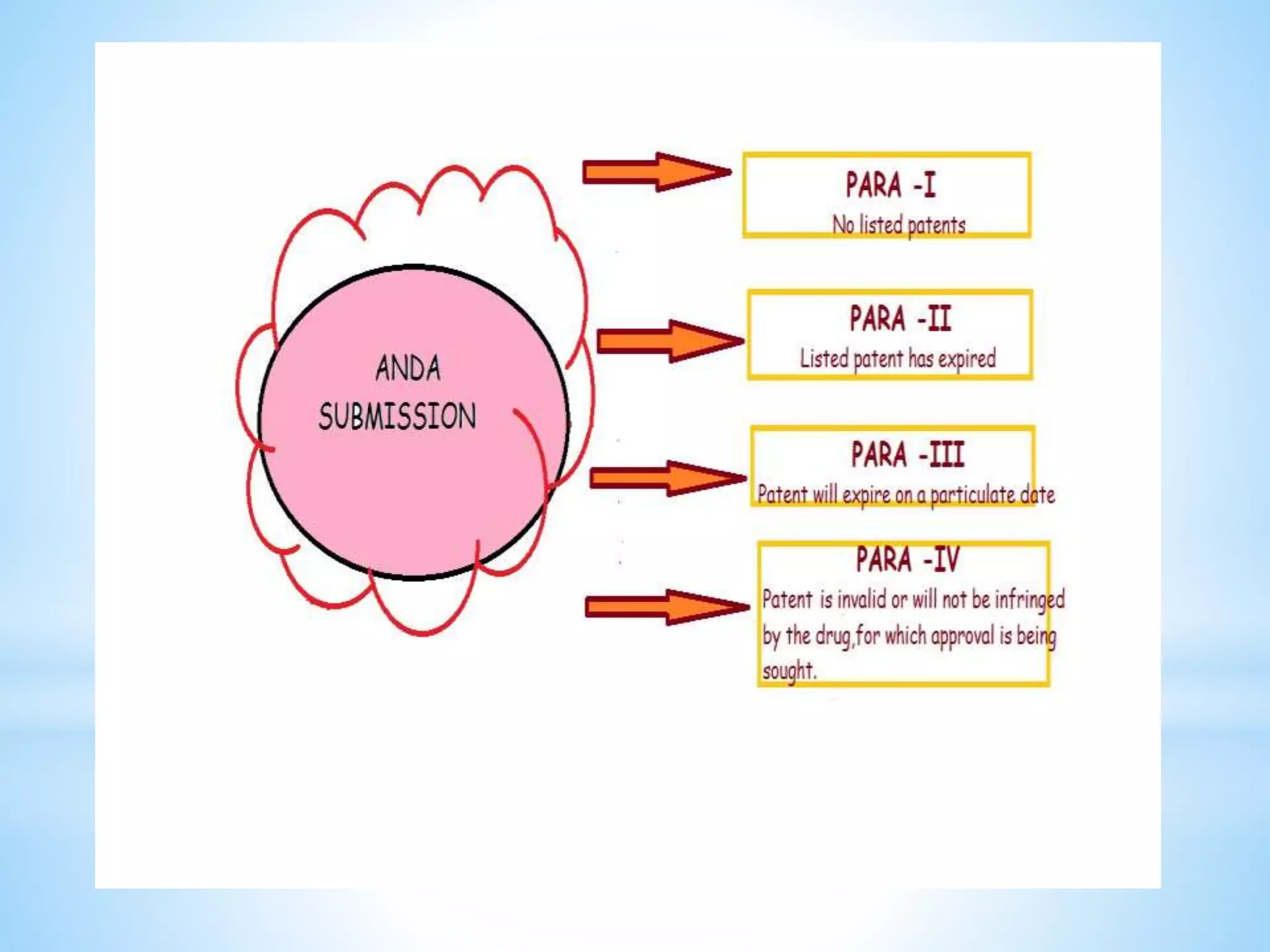
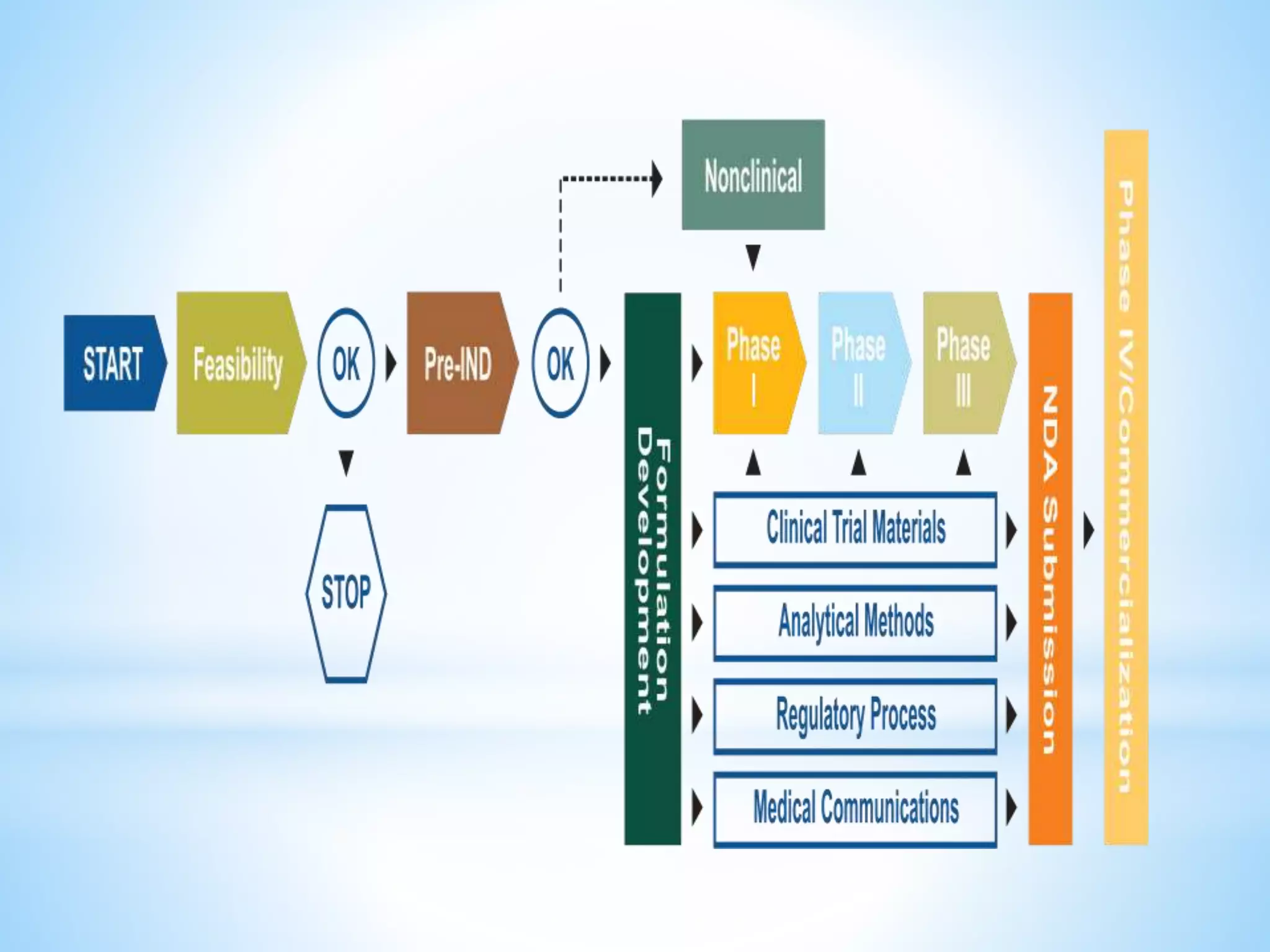

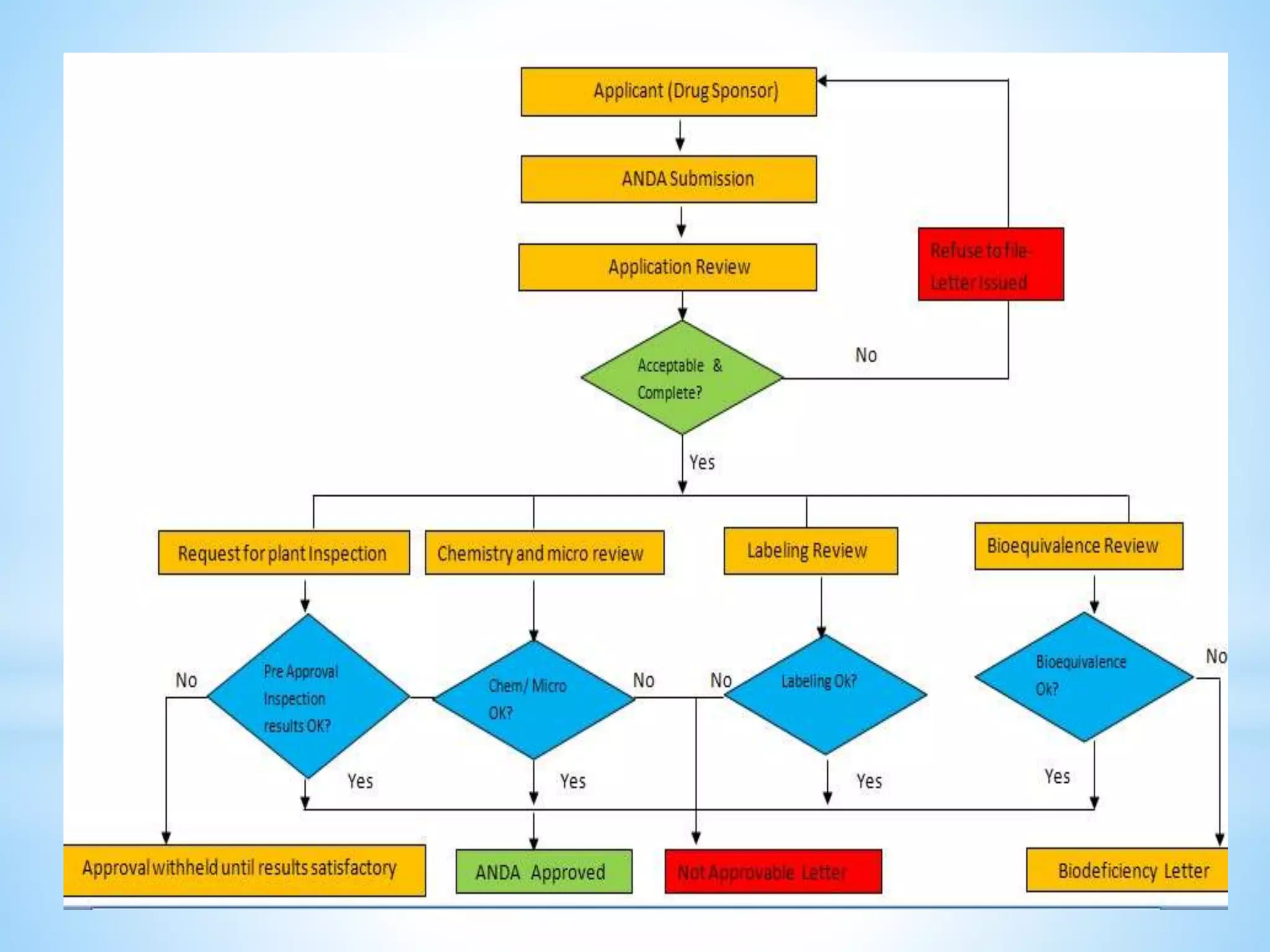

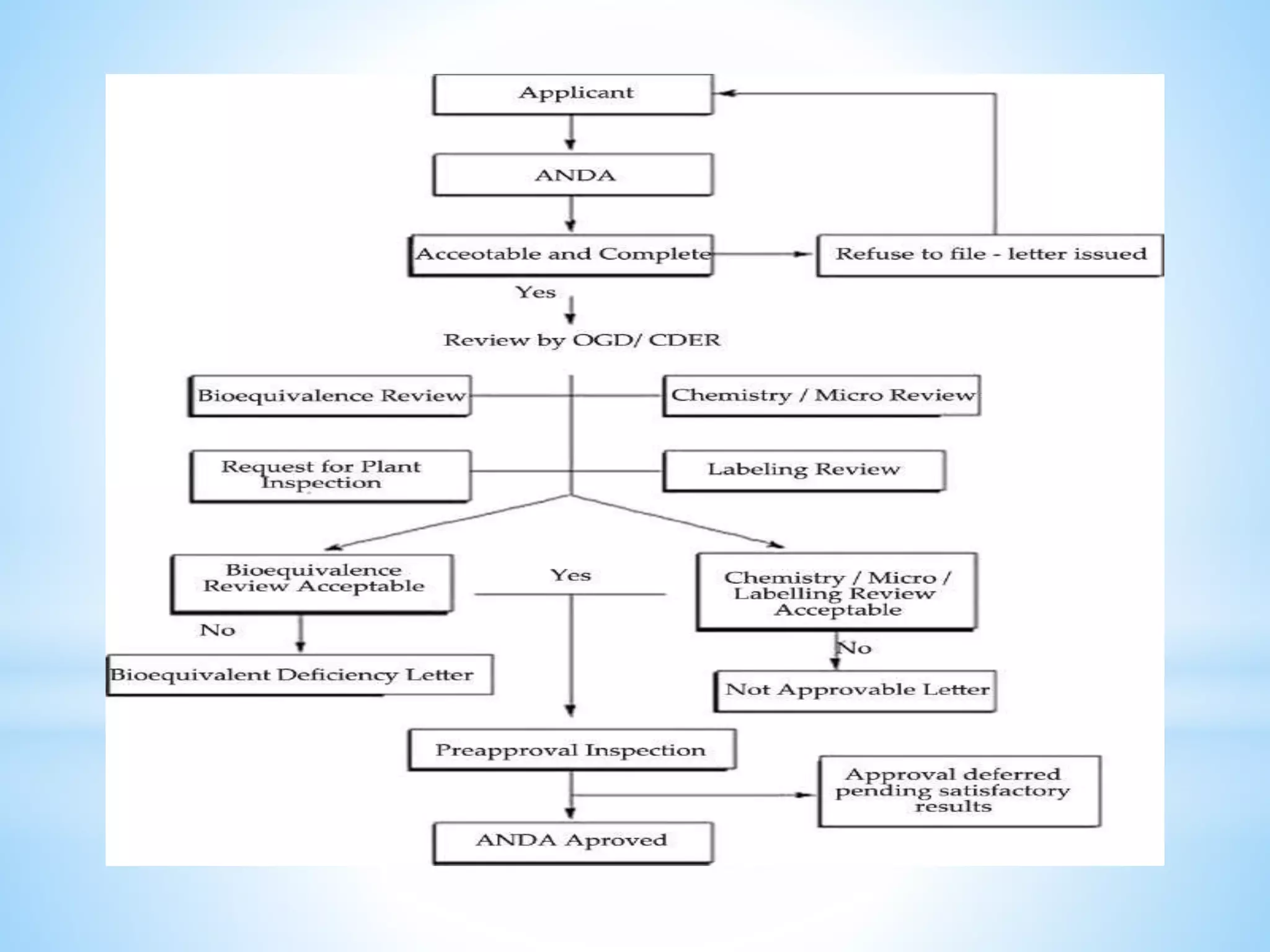
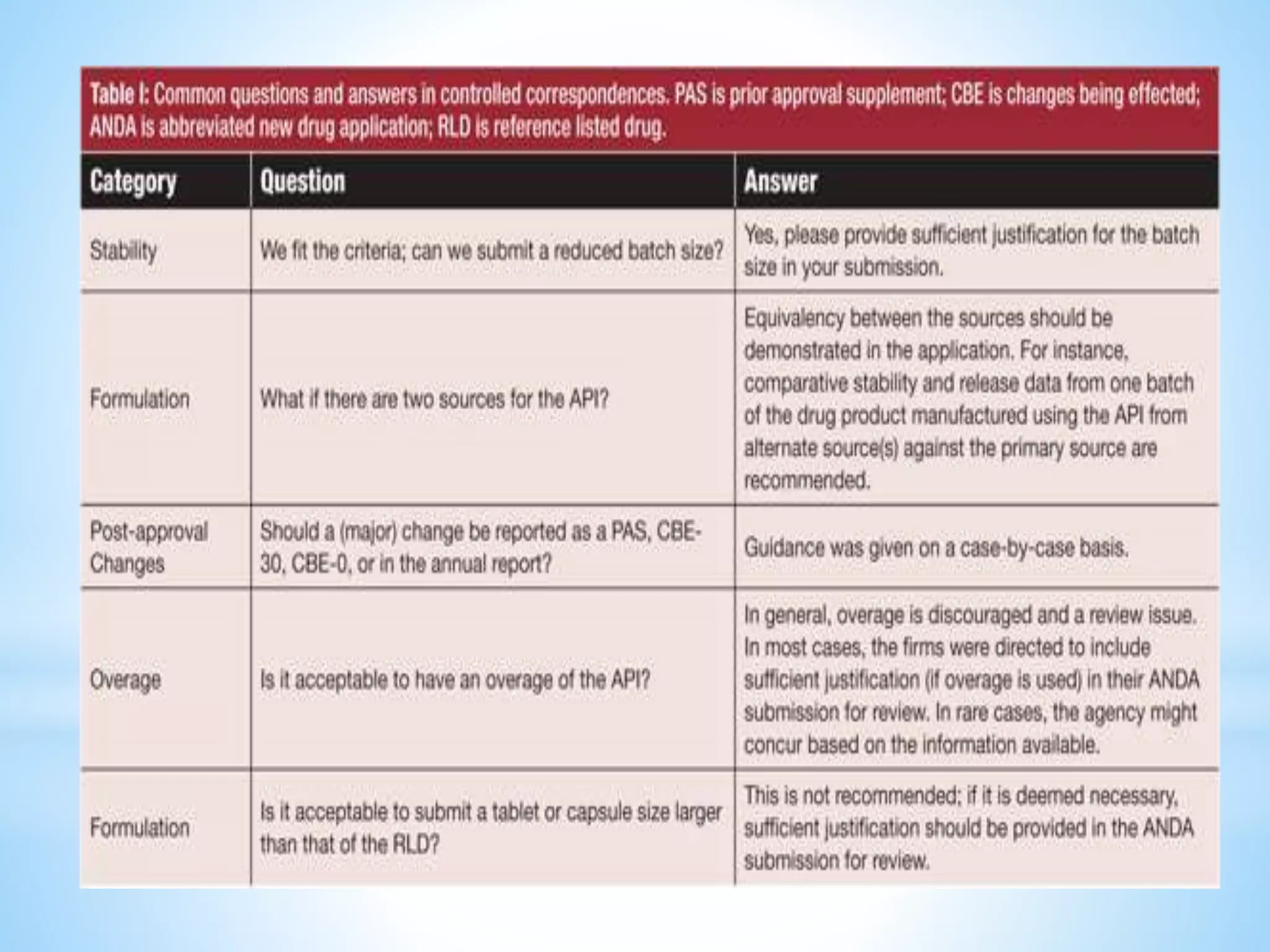


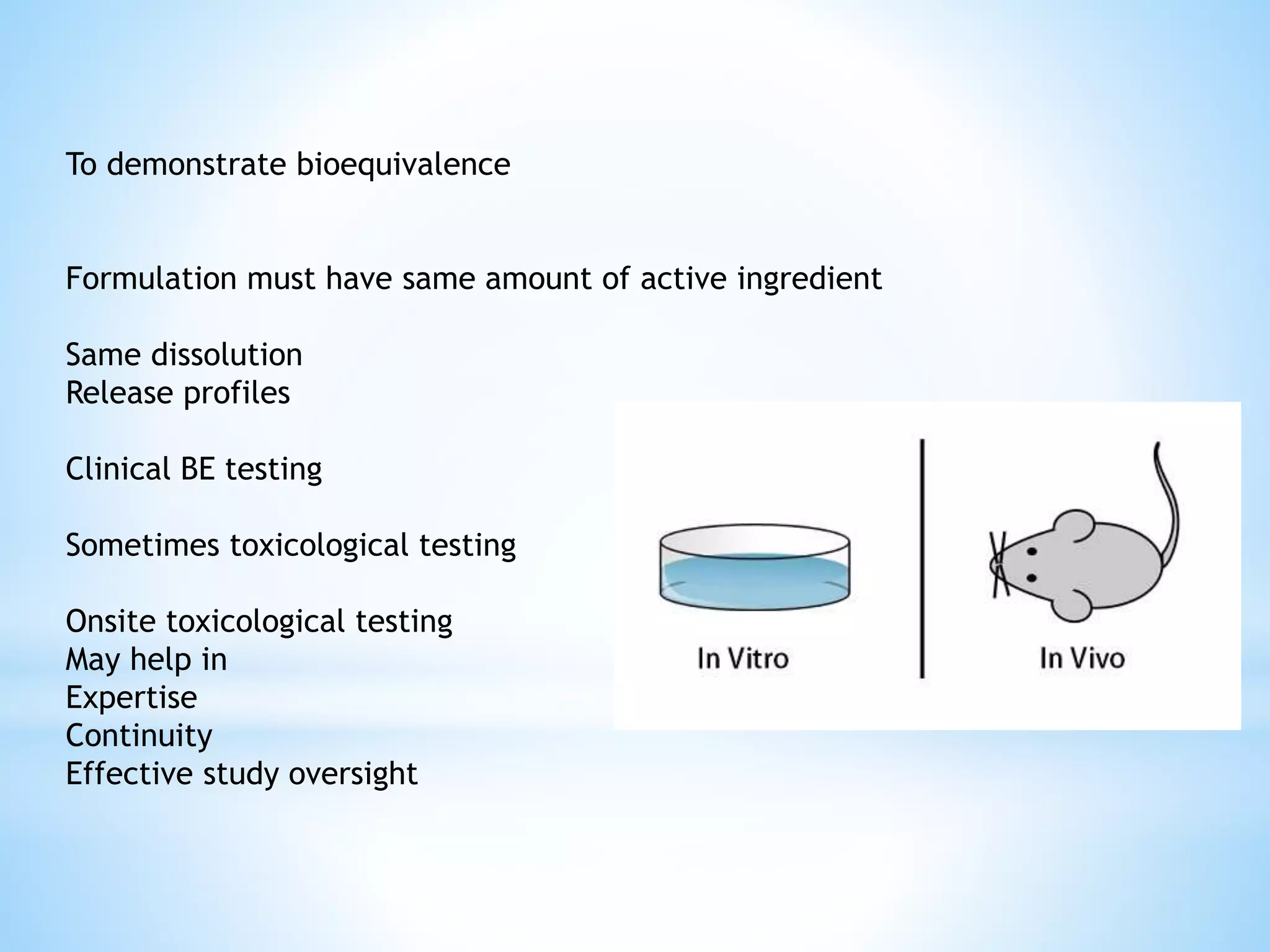
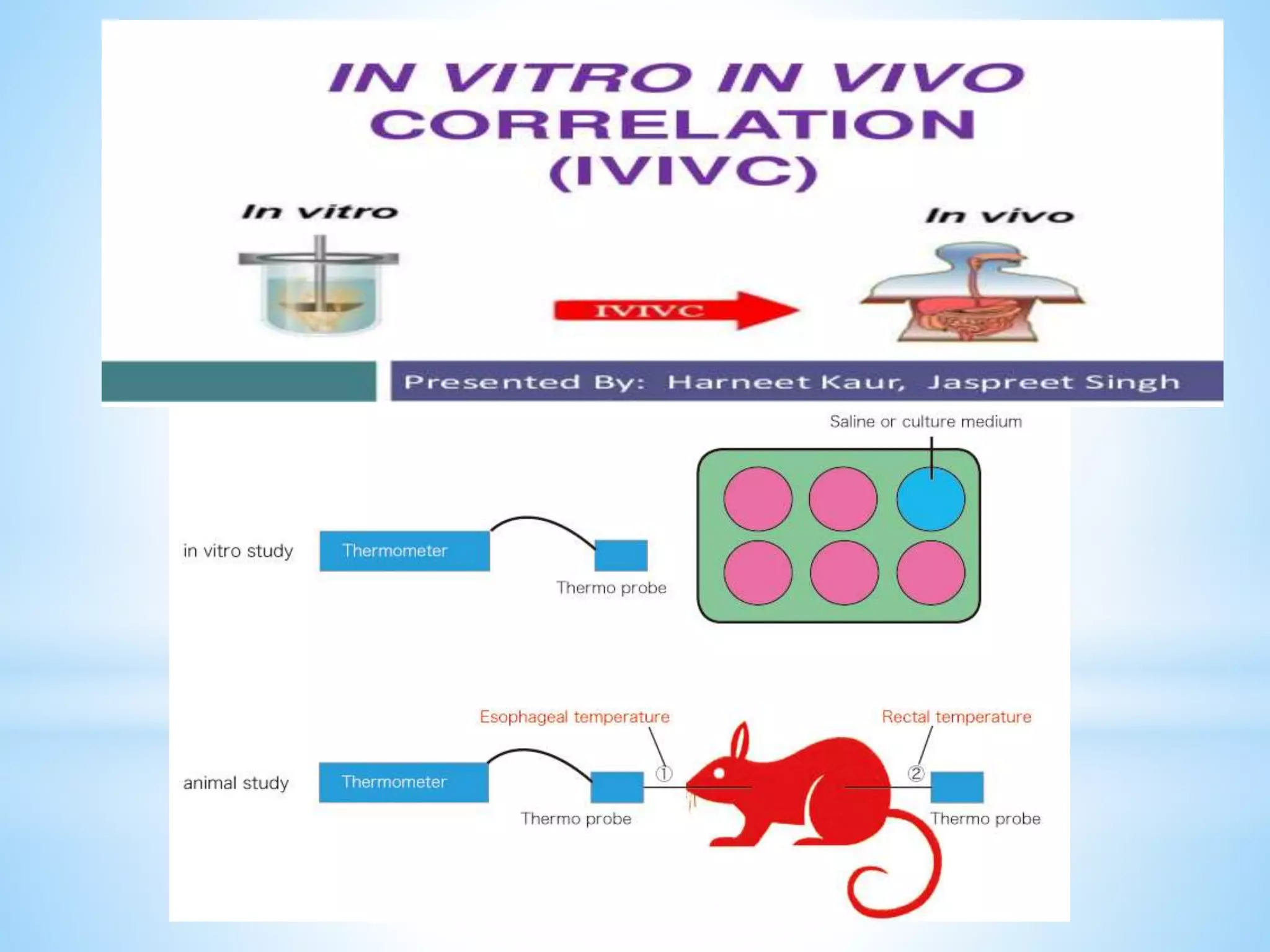

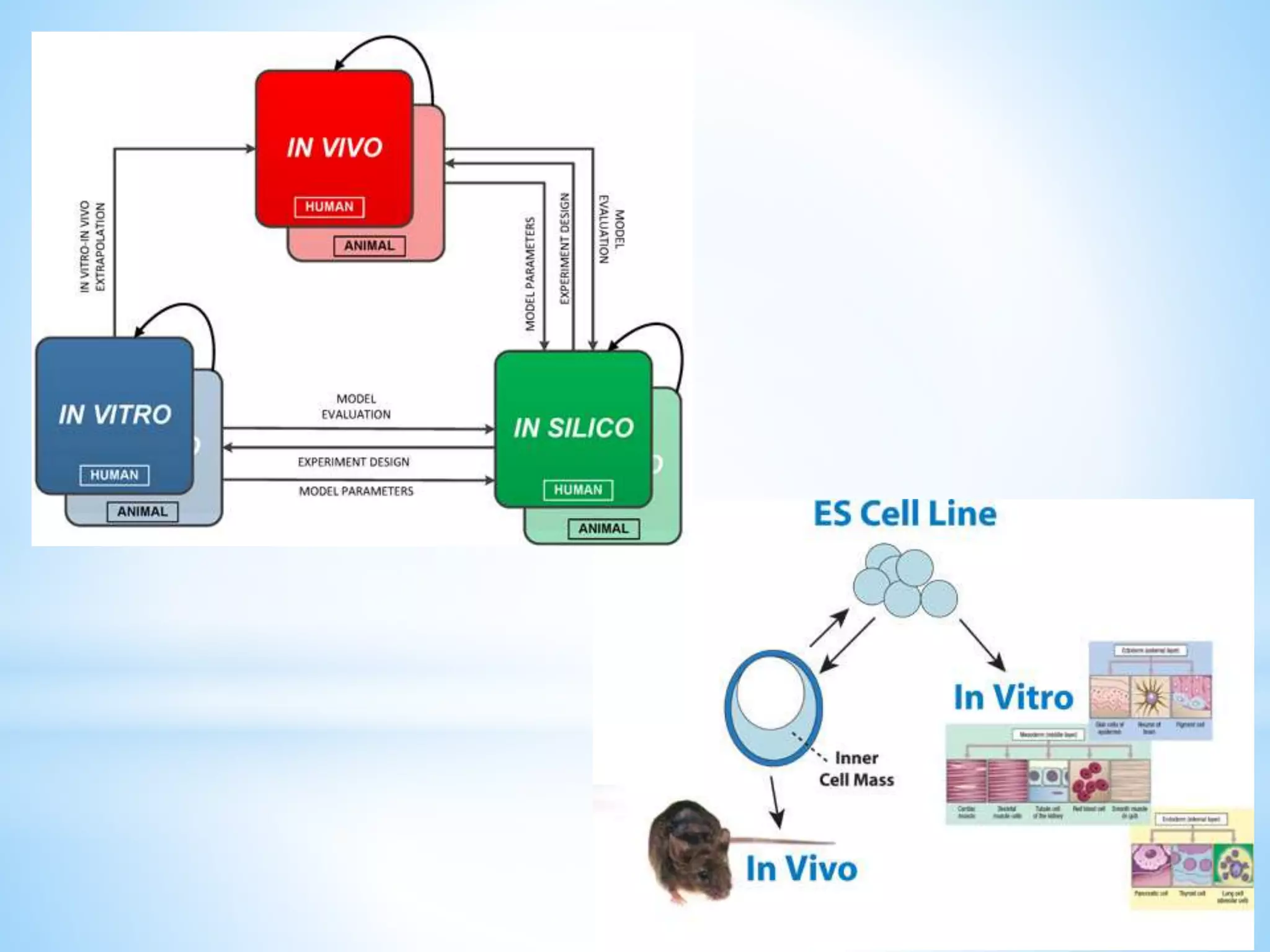
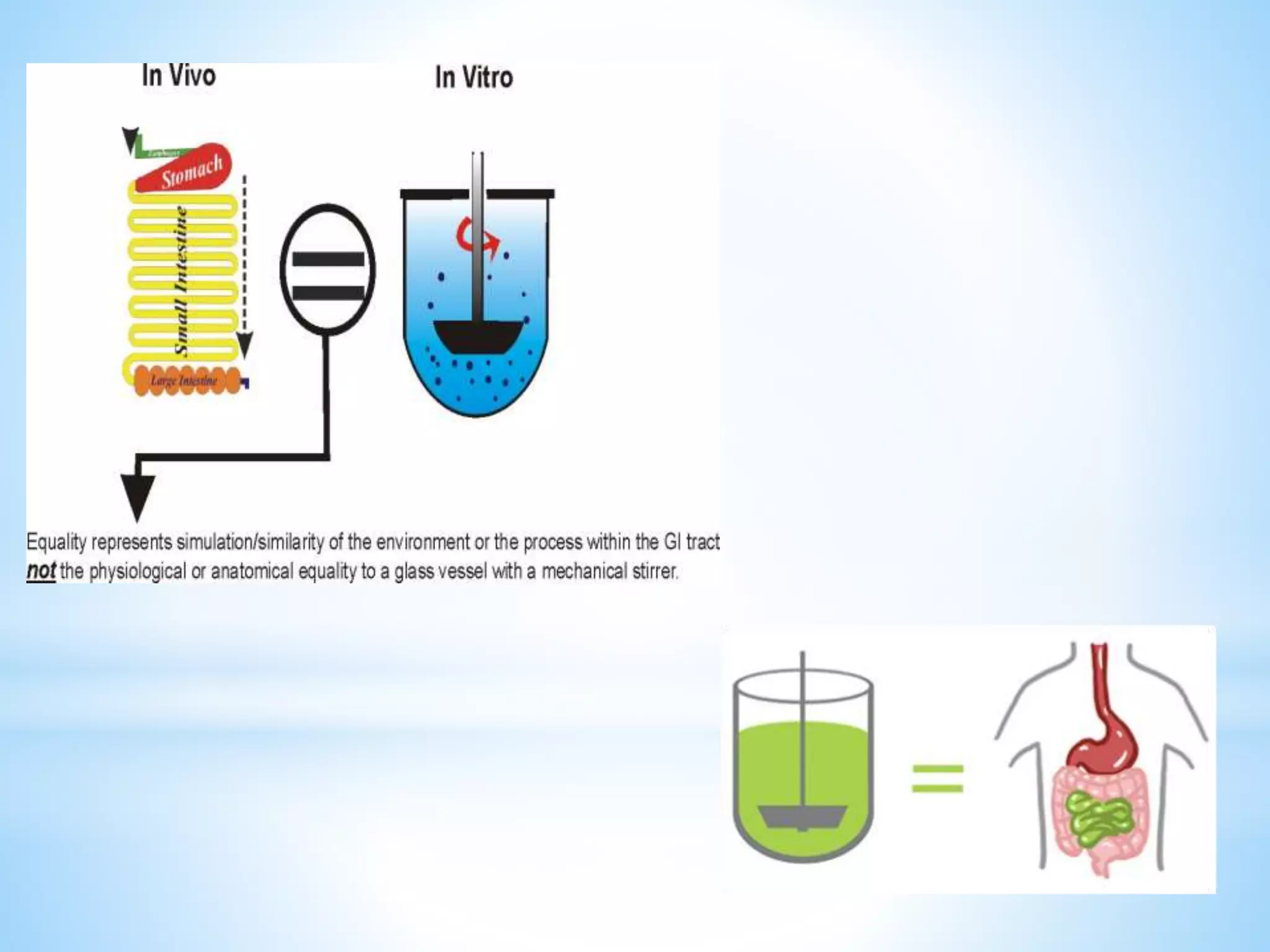

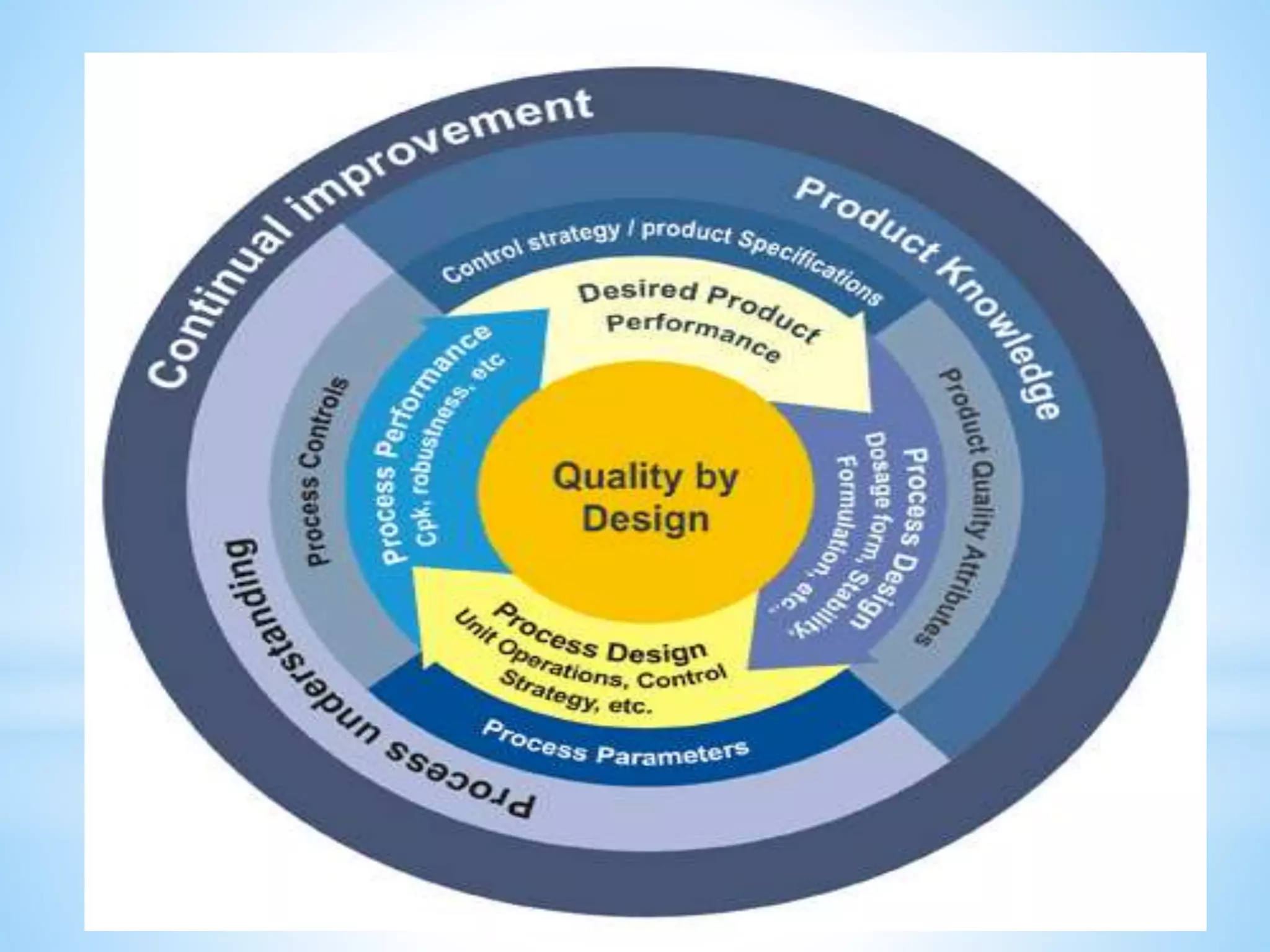
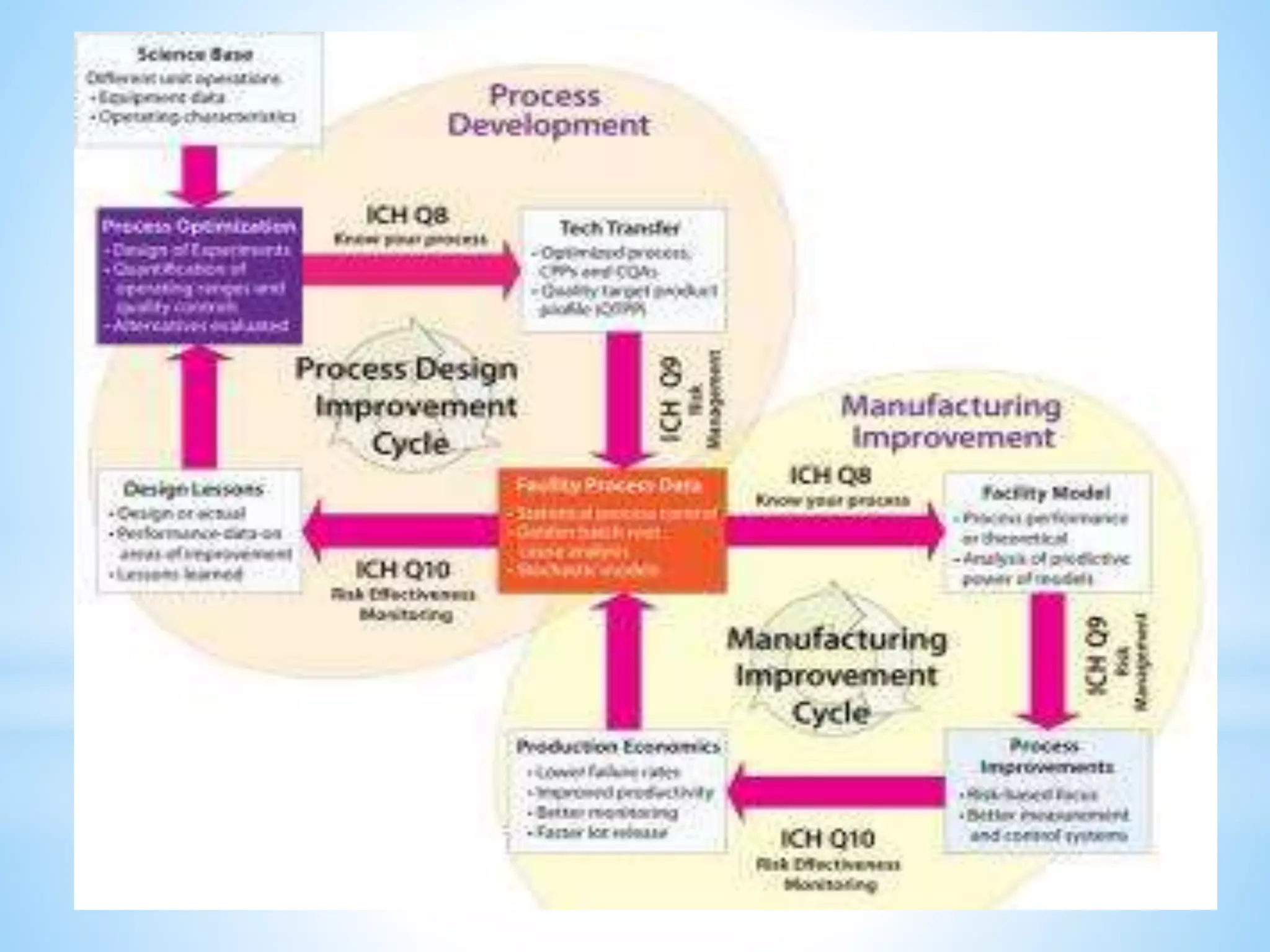

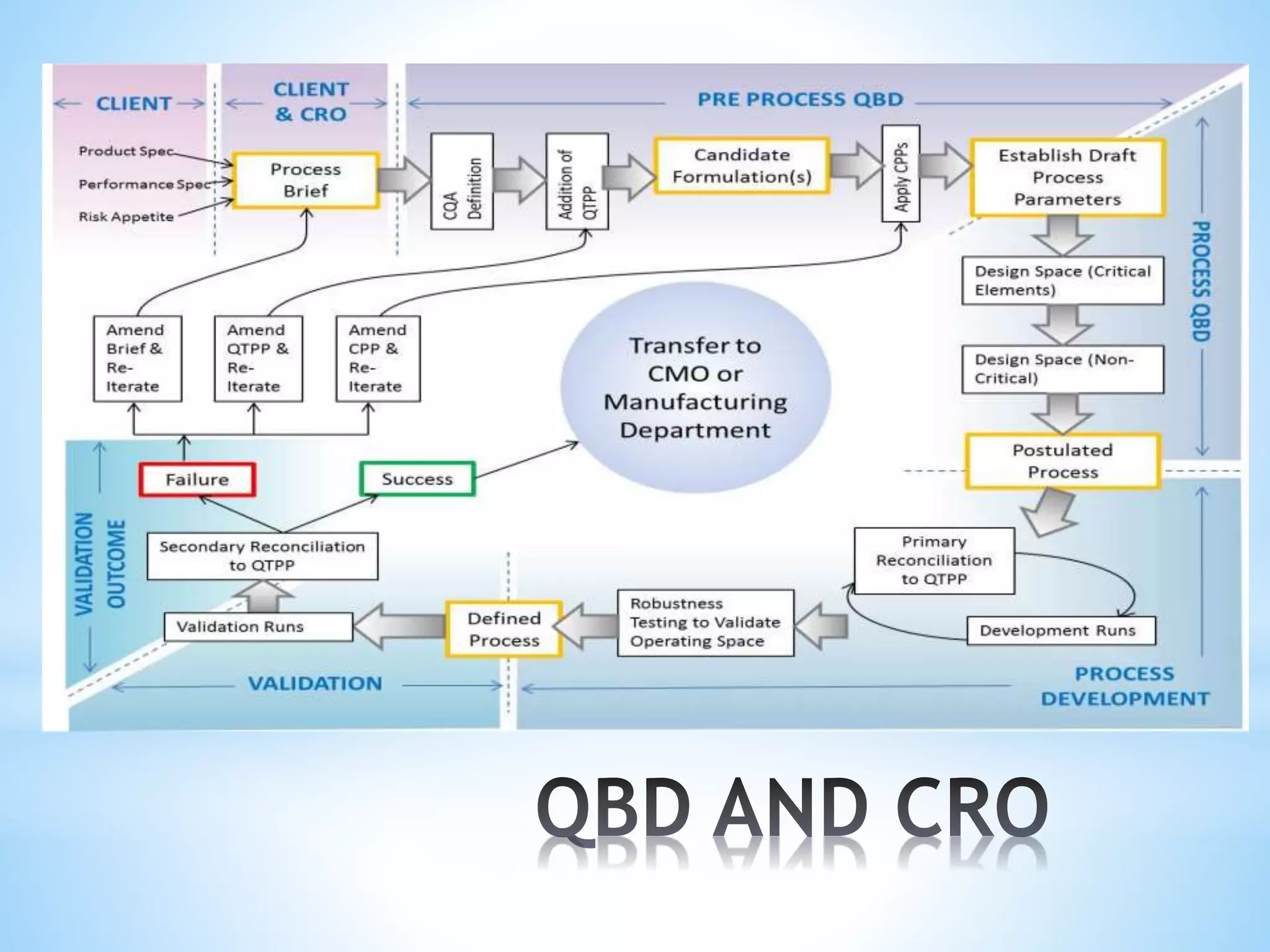






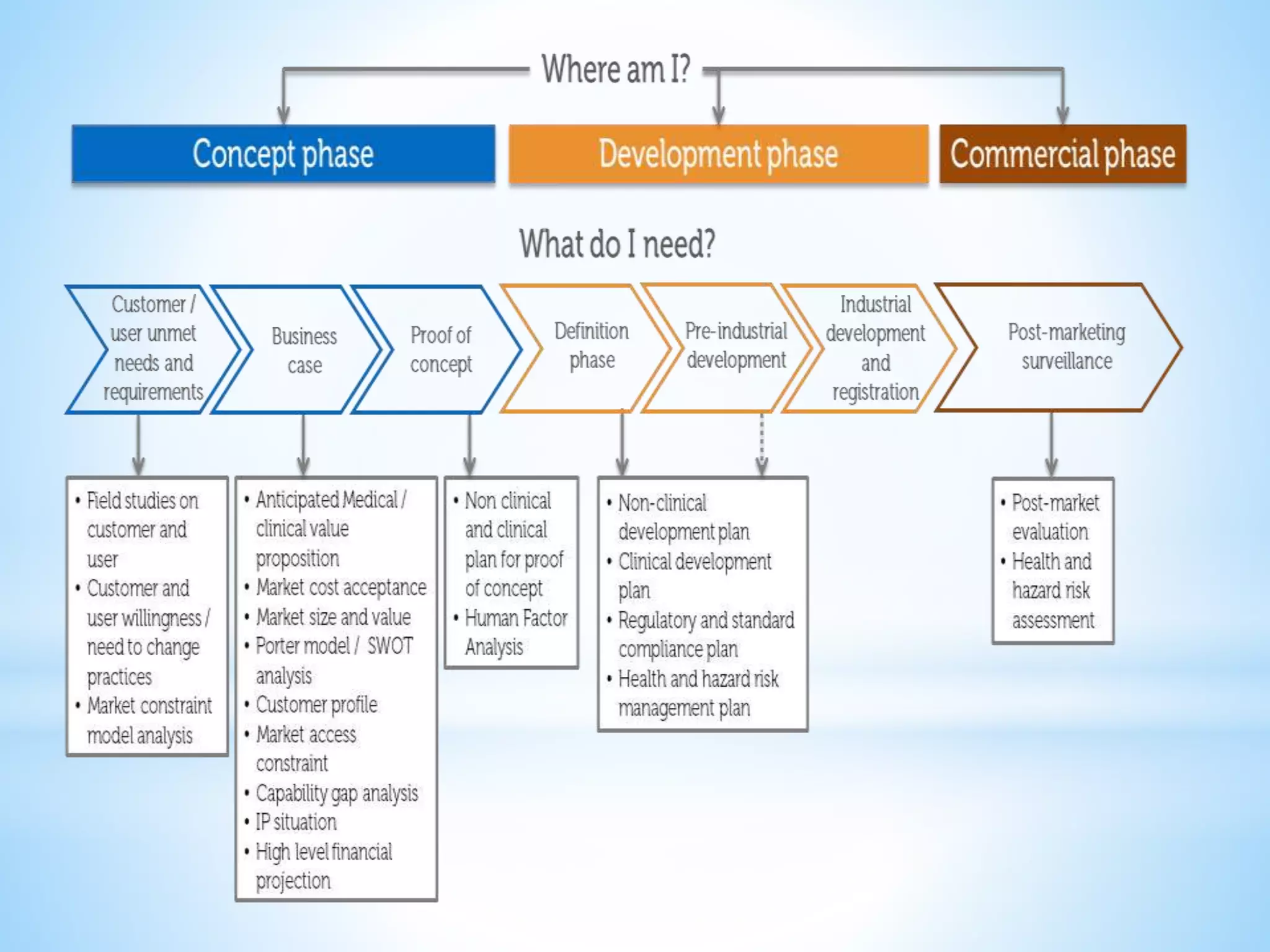
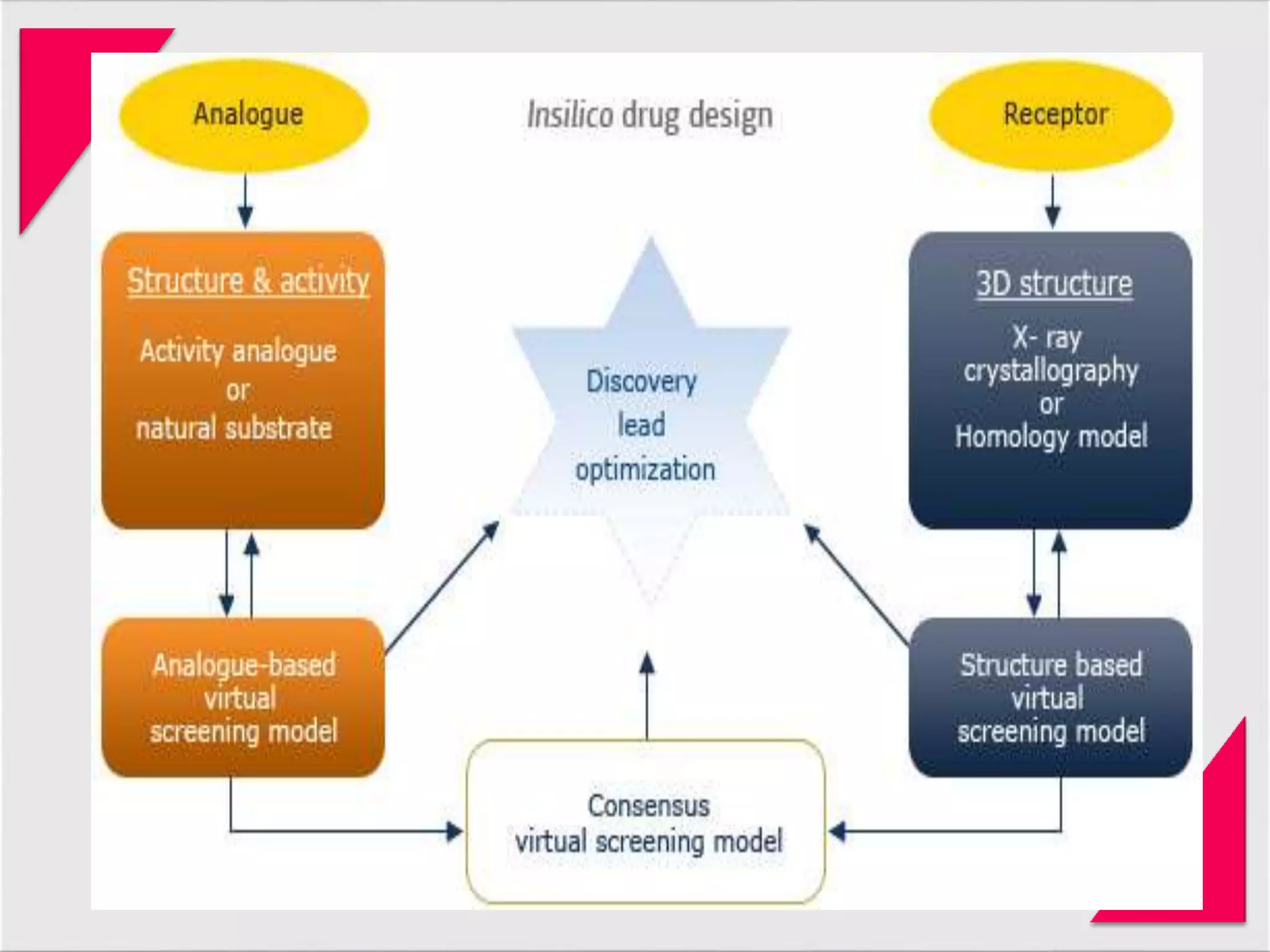
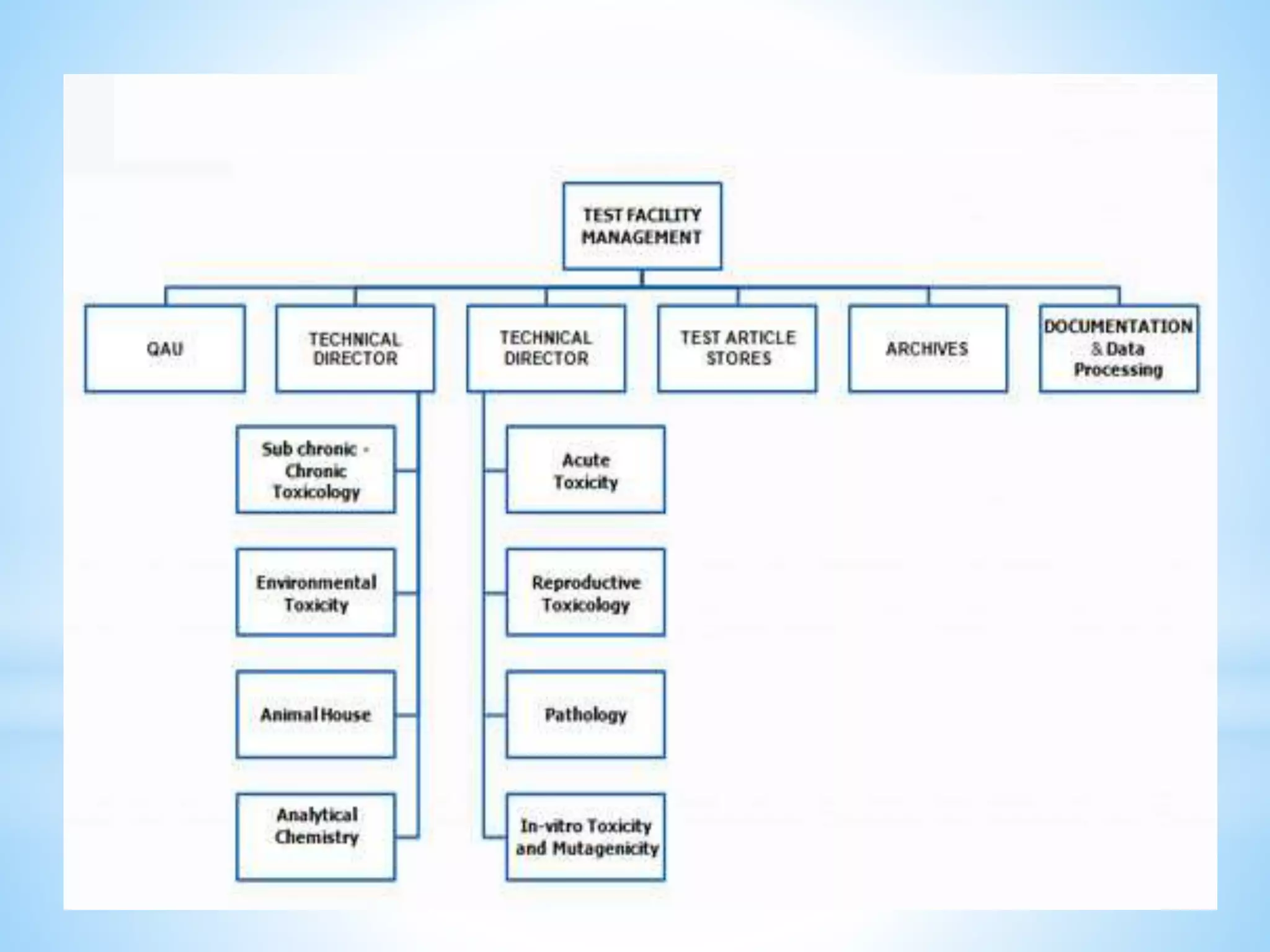
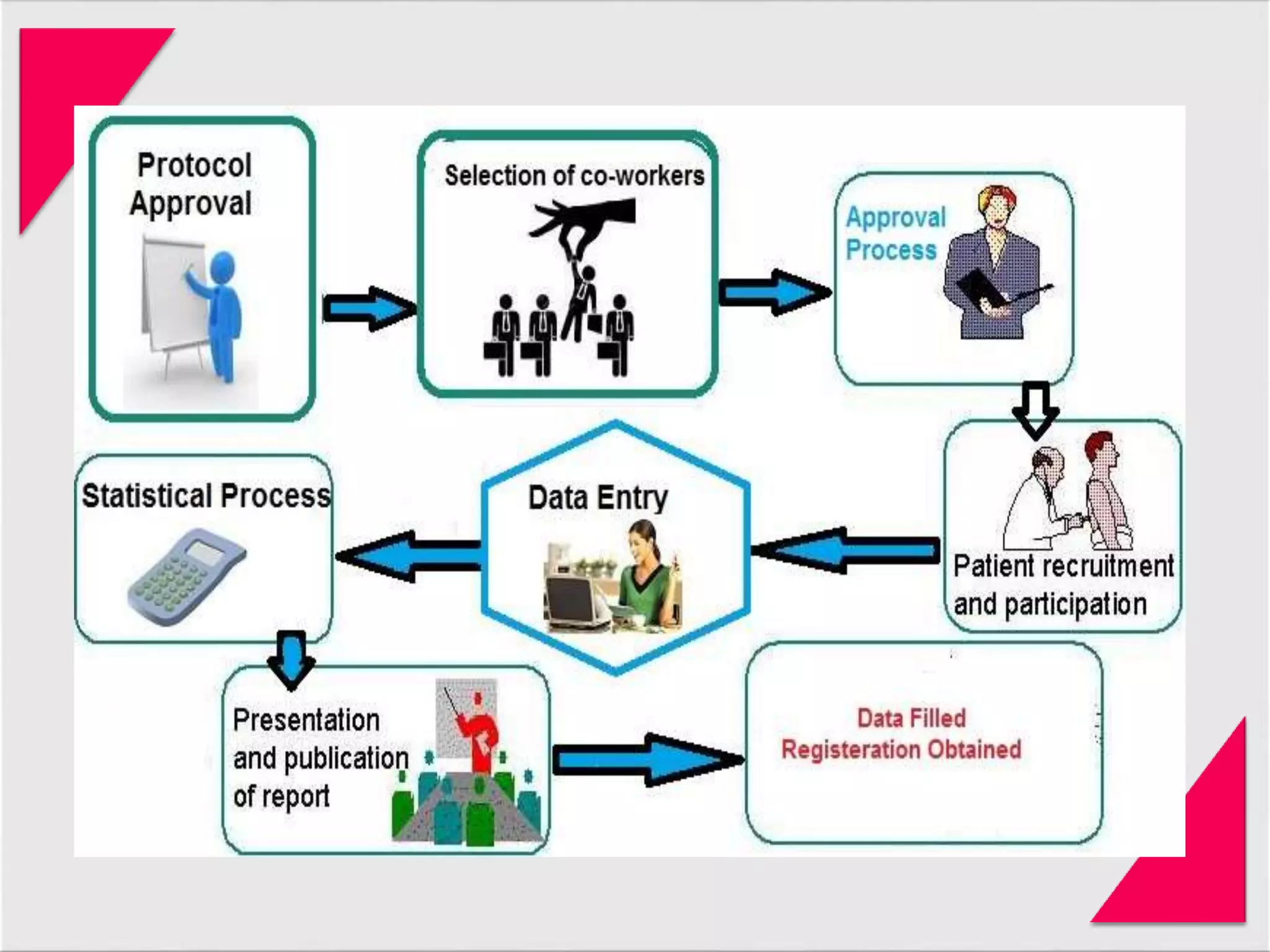
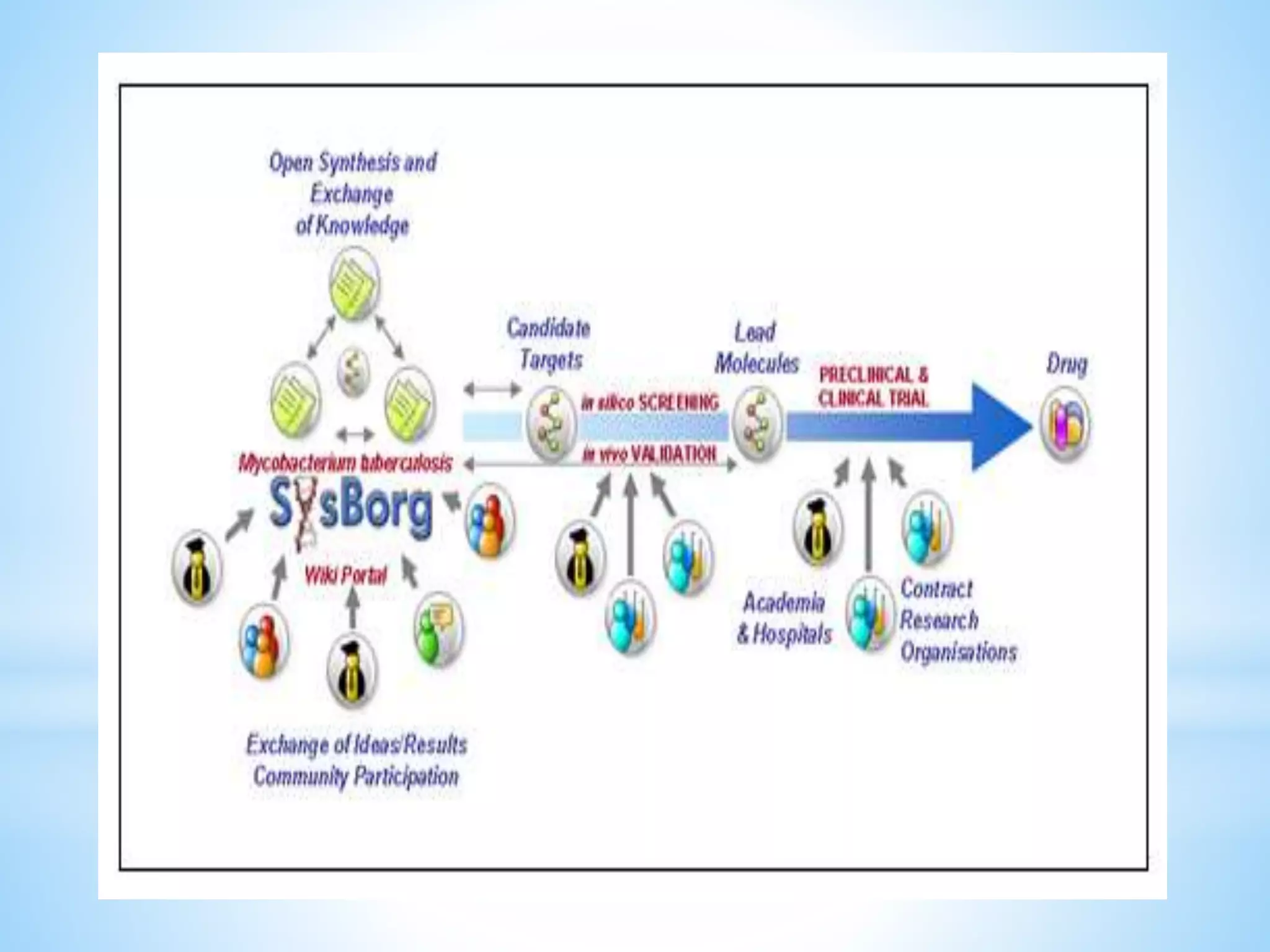
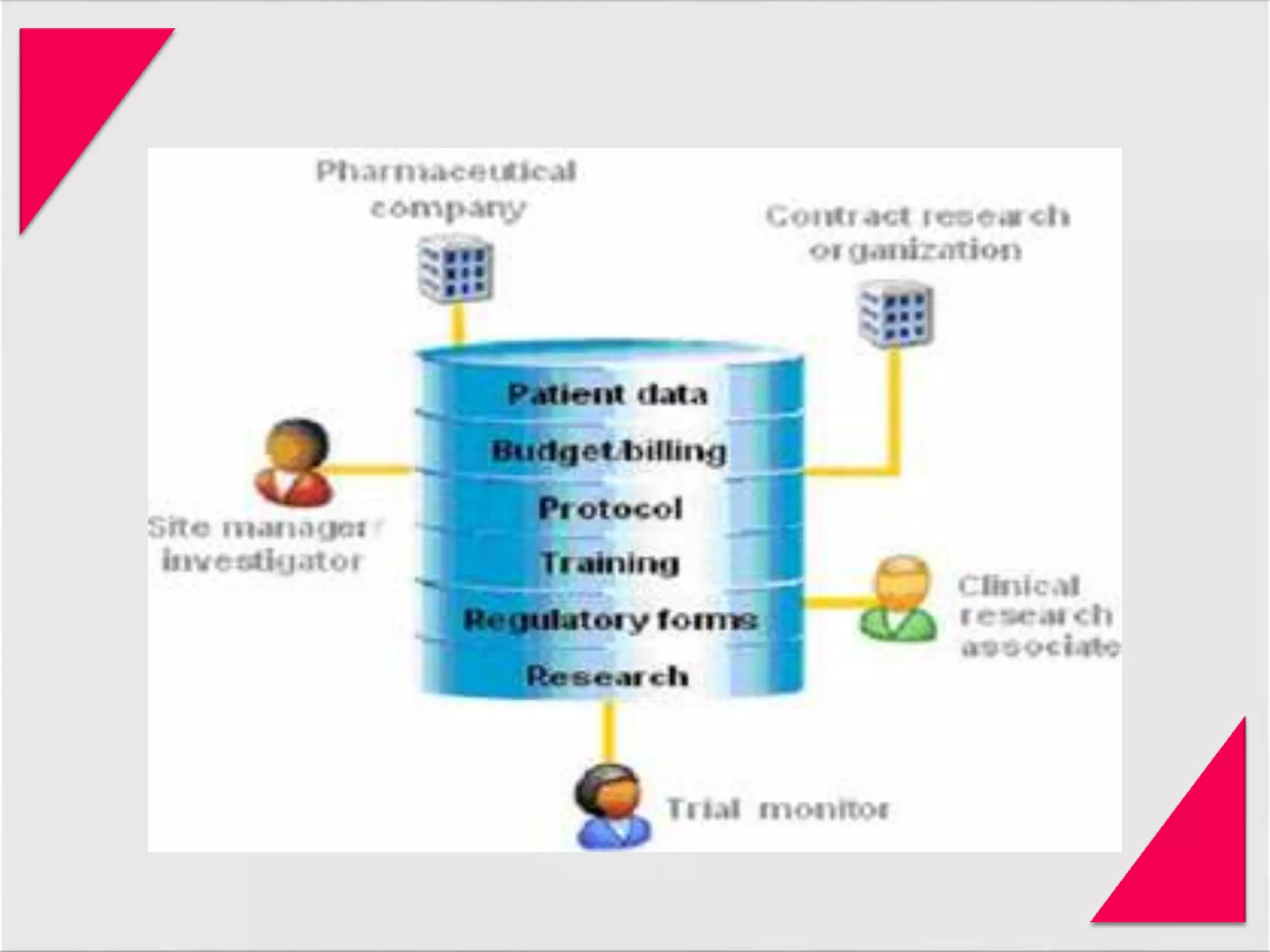



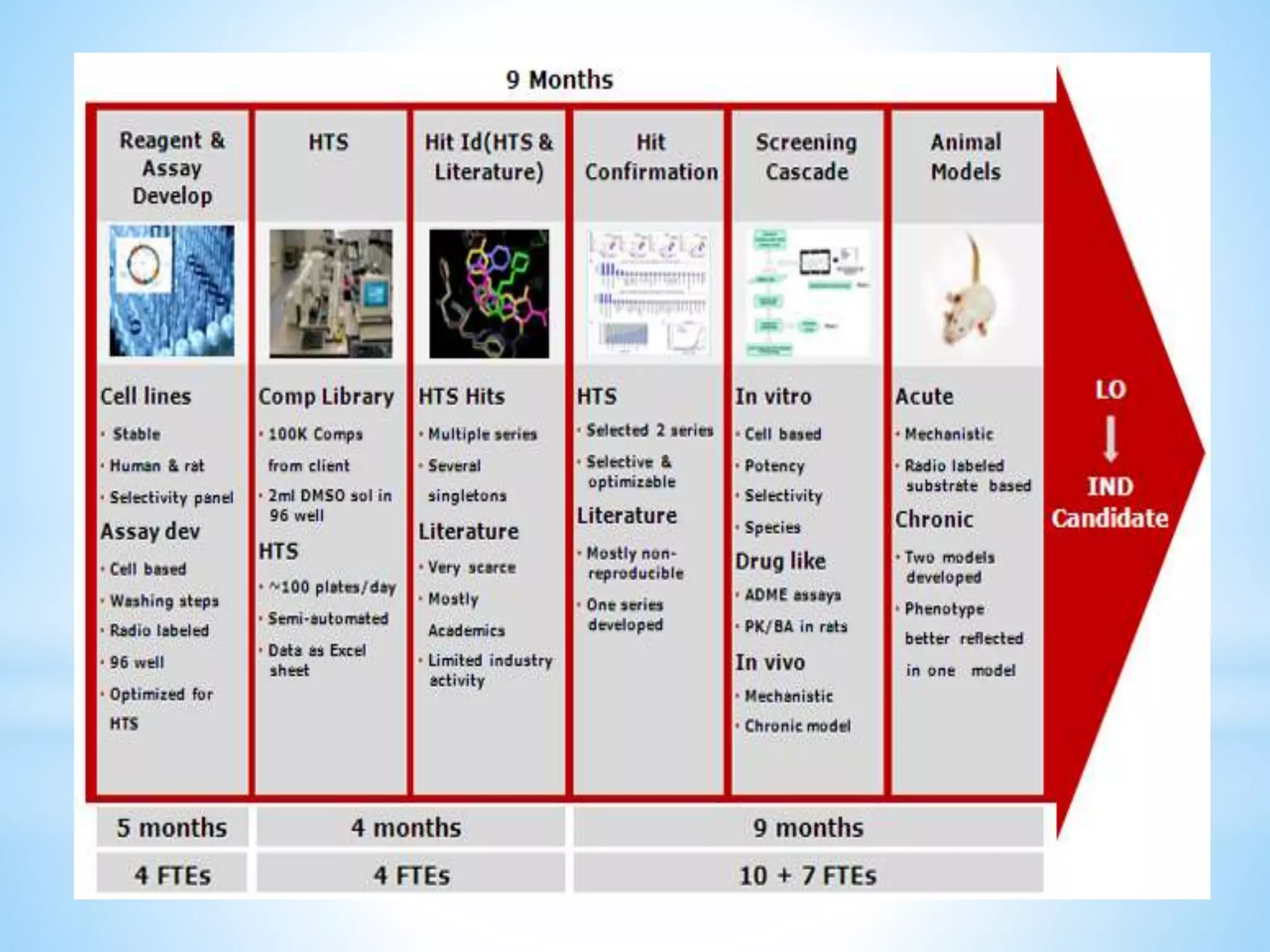
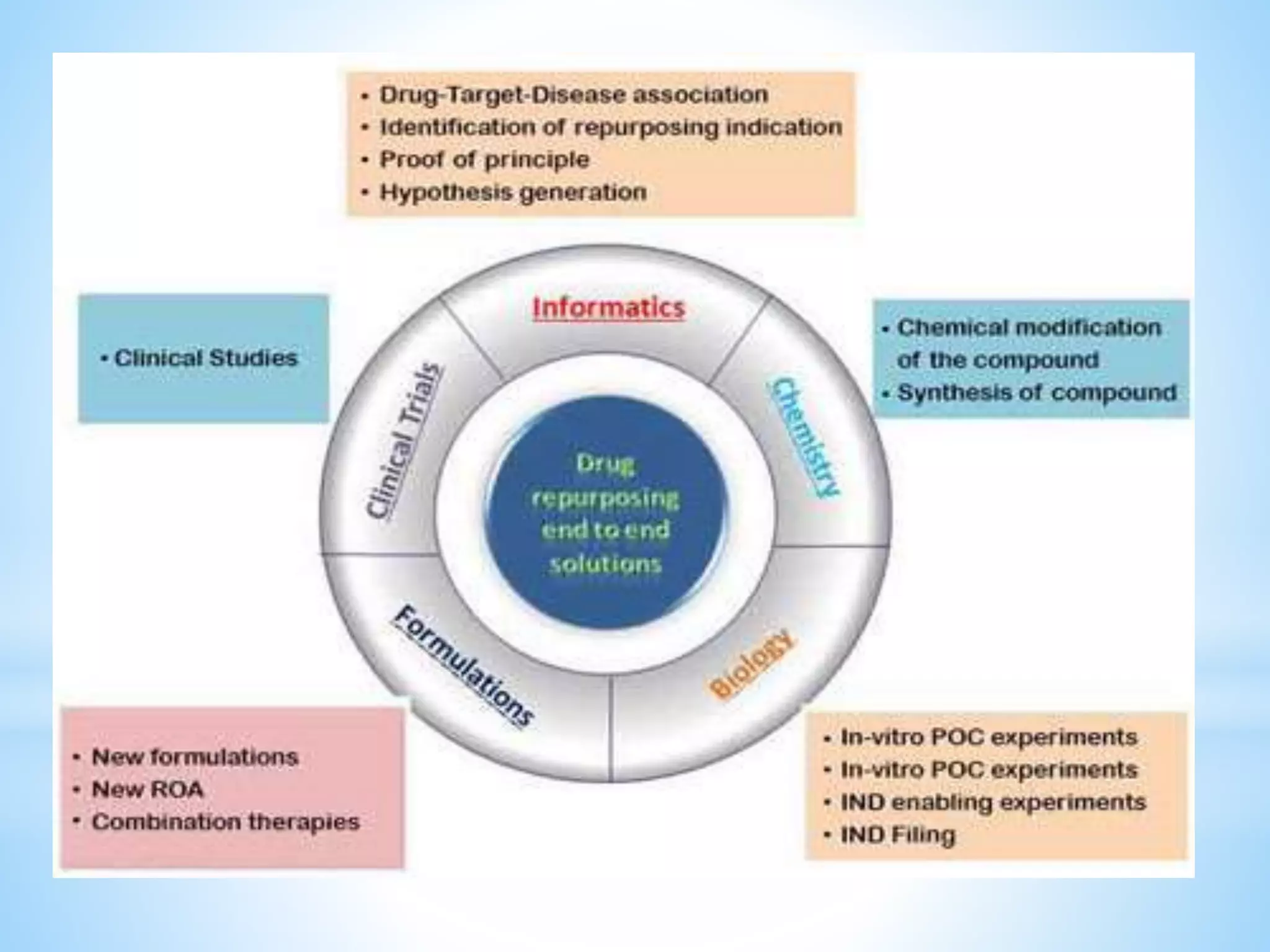

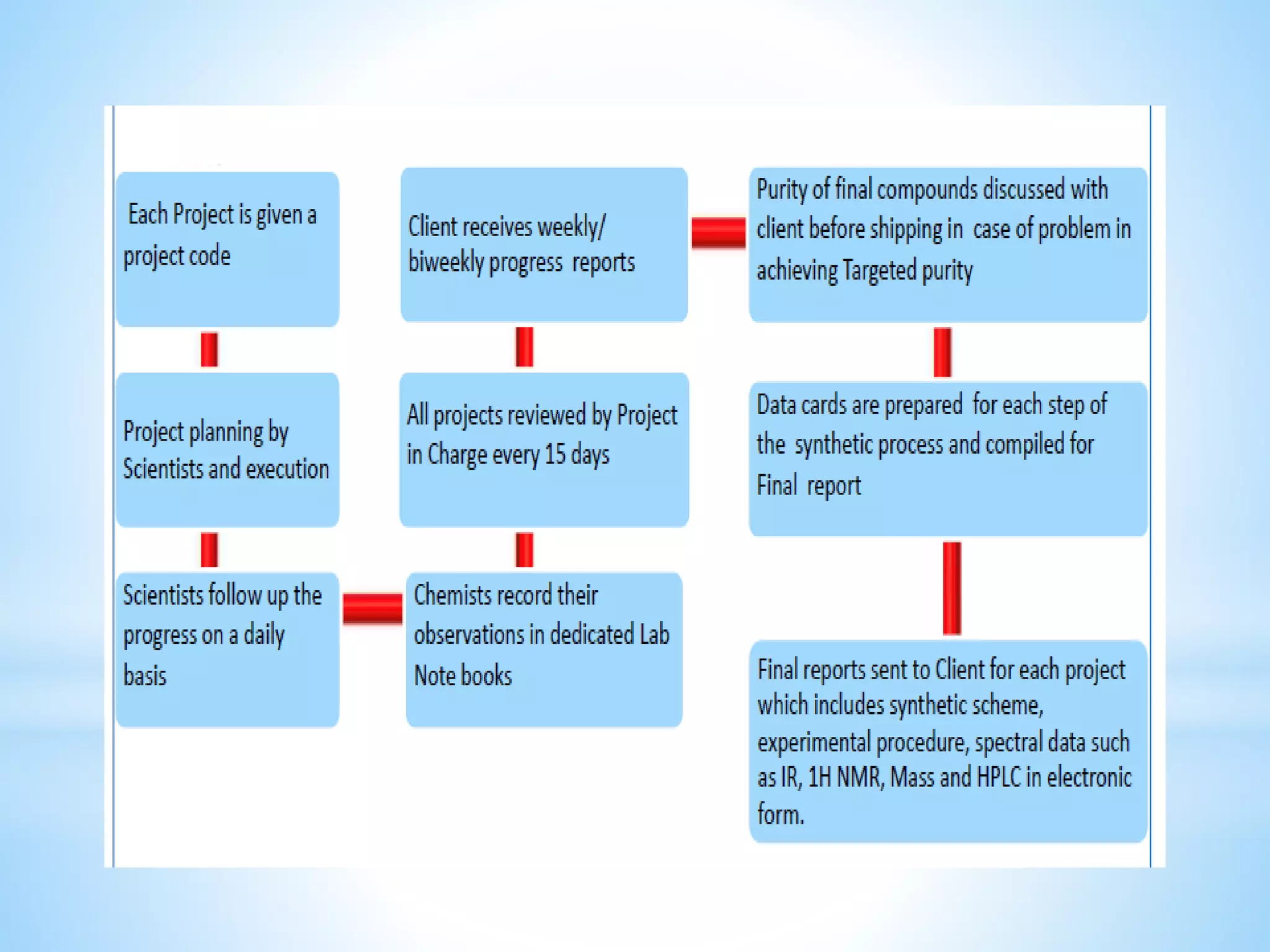
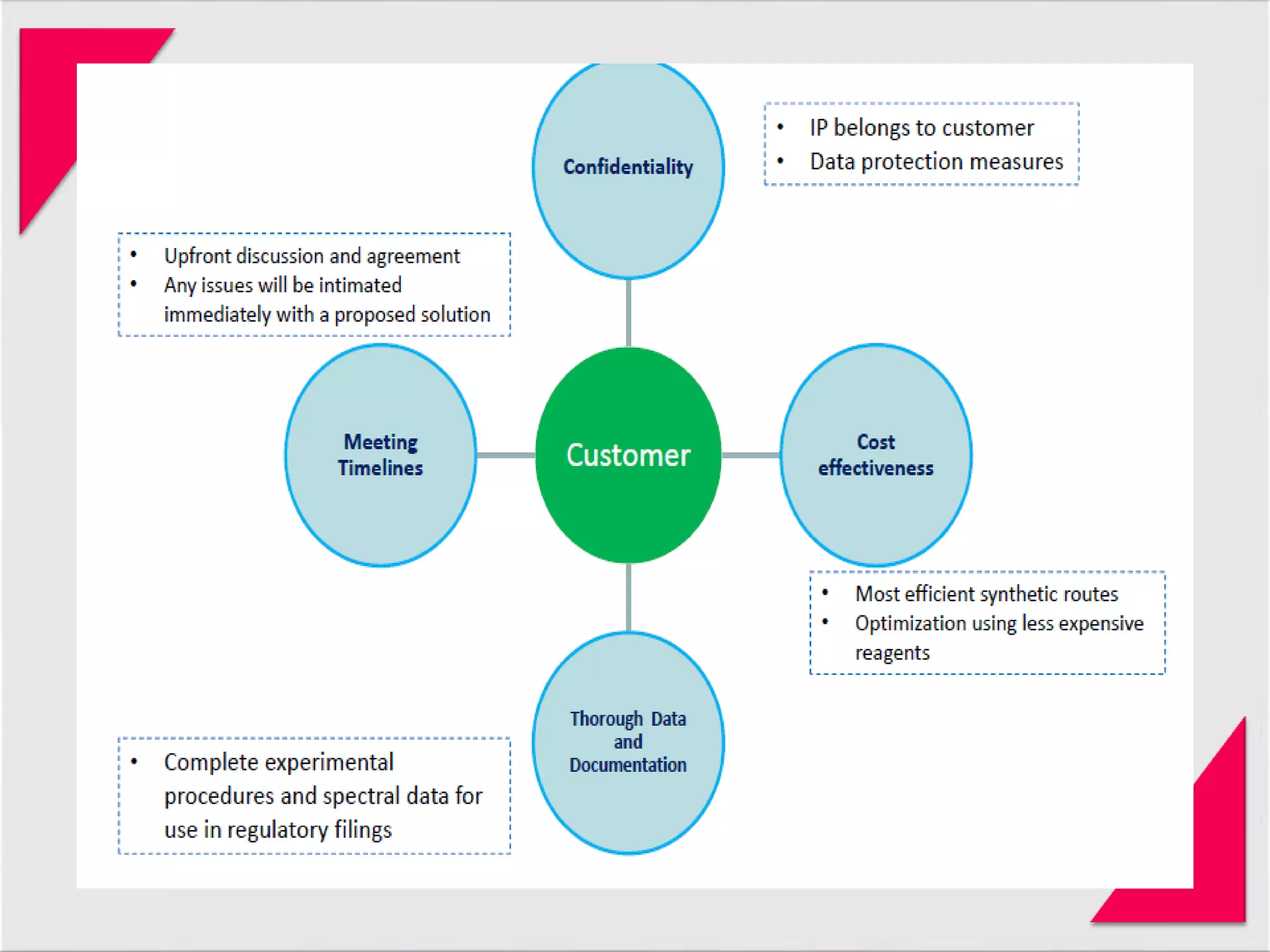











Generic drug companies are relying heavily on qualified contract research organizations (CROs) to accelerate their development processes and be first to market after patents expire on branded drugs. CROs provide expertise across development areas like preclinical research, clinical trials management, bioequivalence studies, analytical testing, and ANDA submissions that help generics meet tight deadlines. Successfully demonstrating bioequivalence through bioavailability and dissolution studies is key for regulatory approval and requires CROs with strong laboratory capabilities and experience navigating regulatory requirements.






















































































