Case record...Postinfectious cerebellitis
•
7 likes•3,770 views

Case record...Postinfectious cerebellitis

Recommended
Recommended
More Related Content
What's hot
What's hot (20)
Viewers also liked
Viewers also liked (10)
Similar to Case record...Postinfectious cerebellitis
Similar to Case record...Postinfectious cerebellitis (20)
More from Professor Yasser Metwally
More from Professor Yasser Metwally (20)
Recently uploaded
Recently uploaded (20)
Case record...Postinfectious cerebellitis
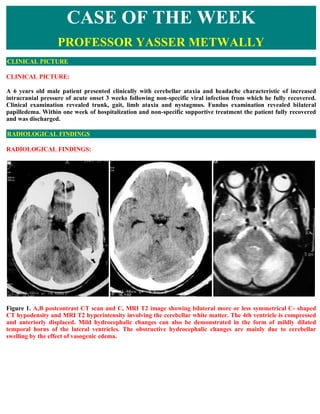
- 1. CASE OF THE WEEK PROFESSOR YASSER METWALLY CLINICAL PICTURE CLINICAL PICTURE: A 6 years old male patient presented clinically with cerebellar ataxia and headache characteristic of increased intracranial pressure of acute onset 3 weeks following non-specific viral infection from which he fully recovered. Clinical examination revealed trunk, gait, limb ataxia and nystagmus. Fundus examination revealed bilateral papilledema. Within one week of hospitalization and non-specific supportive treatment the patient fully recovered and was discharged. RADIOLOGICAL FINDINGS RADIOLOGICAL FINDINGS: Figure 1. A,B postcontrast CT scan and C, MRI T2 image showing bilateral more or less symmetrical C- shaped CT hypodensity and MRI T2 hyperintensity involving the cerebellar white matter. The 4th ventricle is compressed and anteriorly displaced. Mild hydrocephalic changes can also be demonstrated in the form of mildly dilated temporal horns of the lateral ventricles. The obstructive hydrocephalic changes are mainly due to cerebellar swelling by the effect of vasogenic edema.
- 2. Figure 2. MRI T2 images showing bilateral more or less symmetrical C- shaped MRI T2 hyperintensity involving the cerebellar white matter. The 4th ventricle is compressed and anteriorly displaced. Figure 3. MRI FLAIR images showing the bilateral, symmetrical hyperintense C- shaped white matter cerebellar lesions and the mild hydrocephalic changes.
- 3. Figure 4. A, Postmortem section through the cerebellum and the brain stem at the level of the 4th ventricle., B, MRI T2 image at the same level of the postmortem cut. Notice that the MRI T2 hyperintensity is taking the characteristic C- shaped because it is exactly mapping the cerebellar white matter and taking its shape. The MRI T2 C- shaped hyperintensity most probably representing vasogenic white matter edema that is spreading along the white matter tracts and association fibers of the cerebellum. Figure 5. A, Postmortem section through the cerebellum and the brain stem at the level of the 4th ventricle., B, MRI T2 image at the same level of the postmortem cut. Notice that the MRI T2 hyperintensity is taking the characteristic C- shaped because it is exactly mapping the cerebellar white matter and taking its shape. The MRI T2 C- shaped hyperintensity most probably representing vasogenic white matter edema that is spreading along the white matter tracts and association fibers of the cerebellum.
- 4. Figure 6. Same as in figure 4, however the cerebellar white matter color is changes into white to show that the MRI T2 hyperintensity is predominately white matter in location and taking the shape of the cerebellar white matter (C- shaped) and represents vasogenic edema along the cerebellar white matte myelinated axons. In general the characteristic MRI T2 and FLAIR picture of postinfectious cerebellitis is bilateral, symmetrical hyperintensities involving the cerebellar white matter and taking exactly the shape of the cerebellar white matter and mapping it (C-shaped hyperintensity). The C-shape hyperintensity is due to cerebellar white matter vasogenic edema that develops secondary to the immune mediated inflammatory demyelination of the white matter of the cerebellum which results in breakdown of the blood brain barrier with subsequent development of vasogenic edema that follows the myelinated axons of white matter tracts and association fibers of the cerebellum, spreading them apart and extending alongside them resulting in the characteristic C-shape of the MRI T2 and FLAIR hyperintensities. On precontrast CT scan images the bilateral white matter cerebellar lesions appear as a bilateral symmetrical C- shaped hypodensity and on precontrast MRI T1 images the cerebellar white matter lesions appear as a bilateral symmetrical C- shaped hypointensity. Due to breakdown of blood brain barrier, (which is responsible for the formation of vasogenic edema) some degree of contrast enhancement should be expected. Contrast enhancement in the current case appeared linear. Cerebellar swelling (which results from inflammation and edema) might induce compression of the 4th ventricle and variable degrees of obstructive hydrocephalus. Full functional recovery occurred within a week of supportive treatment and MRI examination was normal following recovery and this simply means that the MRI signal changes demonstrated during the acute illness was due to reversible vasogenic edema rather that irreversible structural cerebellar lesions. DIAGNOSIS: DIAGNOSIS: ACUTE POSTINFECTIOUS CEREBELLITIS DISCUSSION
- 5. DISCUSSION: Postinfectious cerebellitis is an example of the benign regressive postinfectious neurological disorders that have the following main characteristics. 1. These disorders are commonly postinfectious of post vaccination in origin (Developing within 5 says to 3-5 weeks after infection or vaccination). 2. They are inflammatory demyelinative white matter diseases in nature characterized pathologically by autoimmune demyelination, breakdown of blood brain barrier with the development of vasogenic edema and contrast enhancement in the acute stage. The MRI signal changes (mainly MRI T2 hyperintensities) observed in these disorders are mainly due to the development of vasogenic edema. 3. They commonly have an acute onset and a regressive course. 4. They commonly have a benign course with good prognosis and full functional recovery should be expected in most cases. This group of neurological disorders are better termed benign regressive postinfectious neurological disorders (BRPIND), and they must be differentiated them from the more malignant and progressive postinfectious neurological disorders such as SSPE (subacute sclerosing panencephalomyelitis) and rubella panencephalitis). Neurological diseases related to the benign regressive postinfectious neurological disorders (BRPIND) are listed in box 1. Box 1. Benign regressive postinfectious neurological disorders (BRPIND) include 1- Postinfectious encephalomyelitis (acute disseminated encephalomyelitis., ADEM) 2- Postinfectious cerebellitis (?variant of acute disseminated encephalomyelitis) 3- Postinfectious transverse myelitis 4- Optic neuritis in children 5- Neuromyelitis optica 6- Guillain-Barré syndrome It is quite apparent that postinfectious neurological disorders have protean clinical presentations depending upon the site{s) involved in the central or peripheral nervous system. Postinfectious neurological disorders might involve the brain and spinal cord (postinfectious encephalomyelitis), the cerebellar only (postinfectious cerebellitis), The spinal cord only (postinfectious acute idiopathic transverse myelitis), The optic nerve only (optic neuritis in children) or the optic nerve and spinal cord (neuromyelitis optica). It is not known whether these disorders represent a single disease with different clinical presentations or different diseases. Pathologically spinal cord involvement in neuromyelitis optica and acute disseminated encephalomyelitis is a transverse myelitic process identical to that of isolated acute idiopathic postinfectious transverse myelitis. Bilateral optic neuritis is characteristically present in acute disseminated encephalomyelitis. It looks like that the division between these postinfectious disorders is indistinct, which is suggestive of a clinical continuum. Benign regressive postinfectious neurological disorders (BRPIND) most commonly occur after smallpox and measles infections. In recent years, the disease has been associated with various viral and bacterial infections.
- 6. Patients may have a history of an exanthem or a nonspecific respiratory or gastrointestinal illness 1 to 3 weeks before onset of neurologic symptoms. Acute cerebellar ataxia (The current case) is a form of acute postinfectious encephalomyelitis following varicella infection. Post-immunization BRPIND occur most frequently following measles, rubella, or mumps vaccination. Many vaccines have been implicated in the causation of BRPIND [Table - 1]. In countries where neural tissue-based vaccines are still used, antirabies immunization with either BPL (betapropionolactone inactivated) or Semple (phenol inactivated) vaccines are important causes for BRPIND. [4] Table 1. Infections alleged to cause BRPIND Viral Measles, Mumps, Varicella, Rubella, Influenza, A,B Hepatitis, A,B Coxsackie, Epstein-Barr, Dengue [16]and HIV [17] Bacterial Mycoplasma pneumoniae, Borrelia burgdorferi, Mycobacterium tuberculosis, Brucella, Chlamydia, Legionella, Salmonella typhi, and Leptospira, Campylobacter, Streptococcus pyogenes Vaccination Rabies, Measles, Rubella, Smallpox, Diphtheria, Mumps, Tetanus, antitoxin, Pertussis, Japanese encephalitis, Polio, Hepatitis B, Influenza, and Meningococcal A, and C Drugs Gold, Arsenical compounds, Sulfonamides, streptomycin/PAS Miscellaneous Allogenic bone marrow transplantation [19], Heart-lung transplantation [19] Herbal extracts [20], Ventriculo-atrial shunts, [21] Stings, Leprosy type I reaction General classification of encephalitis/myelitis (Infectious versus postinfectious) In general encephalitis/myelitis is an acute inflammatory process that affects brain or spinal cord tissue and is almost always accompanied by inflammation of the adjacent meninges. The disease is most commonly caused by viral infection. Encephalitis resulting from viral infection manifests as either acute viral encephalitis or postinfectious encephalomyelitis. Acute viral encephalitis is caused by direct viral infection of neural cells with associated perivascular inflammation and destruction of gray matter. Postinfectious encephalomyelitis follows infection with various viral or bacterial agents; the primary pathologic finding is demyelination of white matter. [1,2,3,16] Postinfectious encephalitis/myelitis is an immunological disorders in which peripheral blood lymphocytes cross- react against myelin basic protein resulting in myelinolysis and inflammatory demyelination of the white matter. Breakdown of the blood brain barrier results in the formation of vasogenic edema that migrate along white matter tracts and is probably responsible for the MRI T2 hyperintensity observed in these disorders. Vasogenic edema is probably responsible for the multisegmental MRI T2 hyperintensity that are commonly seen in postinfectious transverse myelitis that apparently spare gray matter (gray matter is commonly seen as the central dot sign which represents the gray matter squeezed by edema). In postinfectious transverse myelitis vasogenic edema travel up and down along white matter tracts resulting in the multisegmental involvement of the spinal cord that is characteristic of postinfectious transverse myelitis. Pathology and pathogenesis of postinfectious regressive neurological disorders Encephalitis is an acute inflammatory process that affects brain tissue and is almost always accompanied by inflammation of the adjacent meninges. The disease is most commonly caused by viral infection. Encephalitis resulting from viral infection manifests as either acute viral encephalitis or postinfectious encephalomyelitis. Acute viral encephalitis is caused by direct viral infection of neural cells with associated perivascular inflammation and destruction of gray matter. Postinfectious encephalomyelitis follows infection with various viral or bacterial agents; the primary pathologic finding is demyelination of white matter. Direct viral infection of the brain and spinal cord
- 7. involves mainly the gray matter (neurons), while Postinfectious or parainfectious neurological disorders is simply a white matter disease in which there is immune mediated demyelination of the white matter long tracts and the association fibers in the cerebrum, cerebellum, brain stem and spinal cord. Postinfectious neurological disorders might involve the peripheral nerves in Guillain barre syndrome (acute infectious demyelination polyradiculoneuropathy) [16,22,23,24,25,26] The distinction between infective (neuronal) and postinfectious (immune -mediated demyelinative white matter disease) might be difficult or even impossible on clinical background, however table 2 demonstrates the main differences between the two pathologies. Table 2. Differences between infectious and postinfectious encephalitis/myelitis Parameter Infectious Postinfectious Site of involvement Cortical gray Demyelinative, white matter matter disease Mental state Impaired Less impaired, might be clear The interval between the first sign of infection and the onset of Briefer (Few Prolonged (7-21 days) neurological disorders days) CSF examination Abnormal May be normal Postinfectious regressive demyelinative white matter diseases (BRPIND) are characterized by perivenular inflammation and demyelination of brain/spinal cord tissue. In this disorder, peripheral blood lymphocytes cross- react against myelin basic protein. Before widespread vaccination, postinfectious encephalomyelitis most commonly occurred after smallpox and measles infections. In recent years, the disease has been associated with various viral and bacterial infections. Patients may have a history of an exanthem or a nonspecific respiratory or gastrointestinal illness 1 to 3 weeks before onset of neurologic symptoms. Acute cerebellar ataxia is a form of acute postinfectious encephalomyelitis following varicella infection. [16,17,18,26] Postinfectious regressive demyelinative white matter diseases represent an autoimmune response to proteins, most probably myelin- basic proteins, in the CNS with perivenous inflammation and demyelination found in autopsy and biopsy studies. Demyelination may not be present in the first few days of the disease [17]. The strongest evidence for the auto- immune nature of postinfectious neurological disorders is that a similar pathology is seen in experimental allergic encephalitis (EAE). EAE is induced in animals by inoculating them with brain tissue or myelin basic protein. Although EAE now is used as an experimental model for ADEM, the occurrence of ADEM in afflicted humans exposed to rabies vaccine contaminated with brain tissue proves the validity of this model [18] . Further support for the autoimmune nature of ADEM comes from the reactivity of T cells against myelin basic protein found in children with ADEM [19-221 and the increase in proinflammatory cytokines and anti-inflammatory cytokines in the cerebrospinal fluid (CSF) of children with ADEM, even in the absence of ongoing infection [ 23]. Even in the laboratory model of EAE, what is found in one strain of animals does not always apply to others [24]. Autoimmunity can be triggered by several mechanisms, including molecular mimicry, bystander activation, epitope spreading, and mistaken self [25]. The role of of these different mechanisms is unknown. [30]
- 8. Figure 7. Histopathology studies In ADEM have demonstrated perivenous cuffing with inflammatory cells, especially lymphocytes and macrophages, and loss of myelin Histologically, the acute lesions in BRPIND are characterized by an extensive loss of myelin (perivenous cuffing with inflammatory cells, especially lymphocytes and macrophages, and loss of myelin). This may be in the form of a well-demarcated area of demyelination, although in the acute situation, the edges of the demyelinated lesions often are less well defined, and the demyelination and attendant cellular processes extend into the surrounding rim. Demyelinated fibers may be recognized by an axon devoid of a sheath, as seen histochemically, or immunohistochemically, or on electron microscopy by the presence of naked axons. In addition, thinly myelinated fibers may be seen within the lesion, suggesting partially demyelinated or remyelinated fibers. The presence of oligodendrocytes showing the re-expression of myelination proteins suggests the latter event is occurring in a least a significant number of these fibers. Vasogenic edema (due to breakdown of blood brain barrier) may be severe, and is seen as an expansion of the extracellular space, spreading apart both fibers and cells. [16,17,18,19,29,30] Accompanying the myelin loss is a large infiltrate of foamy or debris- filled macrophages lying in sheets that appear to have replaced the normal neuropil. They also may be around the blood vessels, or infiltrating the more preserved areas of tissue as single cells. Depending on the age of the lesion, the macrophages may contain some or none of the myelin proteins described above, or may be LFB positive. The macrophages will stain for general markers such as KPI but depending on the patient's age, early (MRP14) or late (27ElO) markers also may be present to help date lesions. The inflammatory infiltrate varies, but in most acute cases will be of some significance. Lymphocytes staining with the leukocyte common antigen comprise most cells, although polymorphonuclear leukocytes, eosinophils, plasma cells, and even mast cells have been found, together with less well-characterized monocytes. Although they may be present throughout the tissue, they are particularly prominent around the blood vessels, and
- 9. at times may be so severe as to mimic a vasculitis. Both CD4 helper cells and CD8 suppressor cells may be found in the lesions. In the past, there have been suggestions that CD4 cells predominate in early lesions, with CD8 cells taking over at later stages, but this is variable, and a fixed pattern has not been defined. Many workers also have described the occurrence of gamma-delta lymphocytes in these lesions, and their association with acute phase reactant or stress proteins such as heat shock protein on oligodendrocytes has been well recognized. (MS) [23,24,25,26] Demyelination of the white matter is associated with breakdown of the blood brain barrier and the development of vasogenic edema. Vasogenic edema is the most common type of edema results from local disruption of the blood brain barrier. This leads to extravasation of protein-rich filtrate of plasma into the interstitial space, with subsequent accumulation of vascular fluid. This disruption results from loosening of the tight junctions between endothelial cells, and the neoformation of pinocytic vesicles. Once the barrier is breached, hydrostatic and osmotic forces work together to extravasate intravascular fluid. Once extravasated, fluid is retained outside the vasculature, mostly in the white matter of the brain, and within the bundles of myelinated axons of long tracts and commissural fibers. This is because axons run in parallel bundles of fibres with loose extracellular space (that offer low resistance and facilitates the extension of vasogenic edema along myelinated axons which are spreaded apart by the edema) as opposed to gray matter, which has high cell density and is enmeshed in an interwoven network of connecting fibres that offer high resistance to the formation and spread of edema. By definition, this type of edema is confined to the extracellular space. Vasogenic edema is responsible for the MRI T2 hyperintensity and MRI T1 hypointensity and The MRI T1 contrast enhancement frequently observed in these disorders. [16,30] Immunology The lesions of BRPIND are due to autoimmune-mediated inflammation of the CNS, and the absence of viral or bacterial antigens in the CNS is nearly universal. [23,24,25] T-cells have been shown to play an important role, possibly through molecular mimicry or by nonspecific activation of autoreactive T-cell clones. [3] Interleukin-6 may be associated with proliferation of B-lymphocytes and immunoglobulin G synthesis. [44] Anti-basal ganglia antibodies have been demonstrated in children with classical features of ADEM following streptococcal infection. [16,23,24] A complex interplay between cytokines and adhesion molecules is responsible for the cellular events of inflammatory encephalomyelitis and oligodendrocyte death. An association has been established between ADEM and certain class II HLA alleles, indicating that genetic factors may play a role in immunoregulation and progression from infection/vaccination to ADEM. [16,23,24,25,26,27,28] It is quite apparent that Benign regressive postinfectious neurological disorders (BRPIND) comprises a group of neurological disorders which represent a clinical continuum rather that separate diseases entities. They share a common aetiopathogenic factors (antecedent viral infection that invokes antibodies that cross react with the myelin basic protein of the peripheral and central nervous system). They also share a common pathological picture (demyelination of the white matter of the CNS and demyelinating polyneuropathy) and a common prognosis (They all have a very good prognosis with full functional recovery in most cases). [16,27,28,29] BRPIND or multiple sclerosis? In a patient presenting with neurological dysfunction and MRI showing multiple white matter lesions, the most important differential diagnosis is MS. Distinguishing between ADEM and MS is a diagnostic challenge and has important therapeutic and prognostic implications. There are several clinical, imaging, and laboratory parameters that may be useful to distinguish between the two [Table - 3]. CSF electrophoresis has shown a significant reduction in the beta-1 globulin fraction in patients with MS as compared to those with BRPIND and this may be a potential CSF marker. Features that strongly favor BRPIND include a history of preceding infection, polysymptomatic neurological dysfunction, encephalopathy, grey matter involvement (mainly due to extension of the white matter edema to the nearby neurons in the grey matter rather than due to direct involvement of the grey matter ) on MRI, and absence of oligoclonal bands in CSF. [16,27,28,29] Often, distinction between these two conditions cannot be made with certainty and follow-up with serial MRI may be necessary to establish the diagnosis. [16,27,28,29]
- 10. Table 3. Differences between BRPIND and multiple sclerosis Feature BRPIND MS Onset Abrupt Subacute Triggering events Preceding infection or vaccination in Uncommon 70 % of cases Age group More common in children More common in young adult Temporal profile Monophasic, rarely relapsing Relapsing Clinical features Altered sensorium More common Rare Seizures More common Rare Neurological deficit Multifocal Usually single deficit Optic neuritis Bilateral Unilateral Myelitis Complete, transverse myelitis Partial myelopathy Lower motor neuron signs More common Rare Headache More common Rare Meningismus More common Rare Neuroimaging findings Distribution of the lesions Bilateral extensive lesions Scattered asymmetric lesions White matter lesions Confluent, ill-defined periventricular Well-defined, with periventricular and subcortical white matter lesions preponderance Corpus callosum lesions Rare Common grey matter involvement* Thalamic and basal ganglionic lesions Uncommon are common Edema and mass effect May be present Uncommon Follow-up MRI No new lesions New lesions with dissemination in time and place Cerebrospinal fluid Cell count Mild to moderate pleocytosis Normal to mild pleocytosis Protein Increased Normal Oligoclonal bands Uncommon, transient Common and persistent Mortality 10-25 Uncommon * Mainly due to extension of the white matter edema to the nearby neurons in the grey matter rather than due to direct involvement of the grey matter ACUTE POSTINFECTIOUS CEREBELLITIS The present case is a case of postinfectious cerebellitis (acute cerebellar ataxia) demonstrated by MRI. Cerebellitis is an inflammatory syndrome resulting in acute cerebellar dysfunction, which may occur as a primary infectious, postinfectious, or post-vaccination disorder.[1-11] Also known as acute cerebellar ataxia, cerebellitis occurs most commonly in young children and may be difficult to diagnose on routine clinical and laboratory studies. An encephalitis largely restricted to the cerebellum, called cerebellitis. Cerebellitis may occur due to a host of viral
- 11. agents, including enteroviruses, herpesviruses, HIV, and rabies. Bacterial infections have also been associated with cerebellitis, including Borrelia burgdorferi (Lyme disease), Mycoplasma pneumoniae, Legionella, and Coxiella burnettii (Q fever). In addition, cerebellitis may follow immunizations such as hepatitis, smallpox, and measles vaccination, or may occur without evidence for an antecedent or concurrent factor. In many cases, however, the precise causative agent is not isolated. [1,2,3,4,5] Our patient most likely had an immune-mediated parainfectious or postinfectious viral cerebellitis. He developed an idiopathic acute cerebellar ataxia 2 weeks following non-specific viral infection. The CSF abnormalities and the reversible MRI findings of swelling, parenchymal signal changes, and cerebellar enhancement point to a parainfectious or postinfectious inflammatory etiology. The MRI C-shaped cerebellar white matter edema demonstrated in this patient is mainly due to cerebellar white matter vasogenic edema that totally disappeared on MRI follow-up studies following complete clinical recovery. Any involvement of the cerebellar gray matter in postinfectious cerebellitis is probably secondary to white matter edema and is due to spreading of edema to the nearby neurons. Vasogenic edema fluid is retained outside the vasculature, mostly in the white matter of the brain, and within the bundles of myelinated axons of long tracts and commissural fibers. This is because axons run in parallel bundles of fibres with loose extracellular space (that offer low resistance and facilitates the extension of vasogenic edema along myelinated axons which are spreaded apart by the edema) as opposed to gray matter, which has high cell density and is enmeshed in an interwoven network of connecting fibres that offer high resistance to the formation and spread of edema. Although the cerebellar MRI signal changes are commonly bilateral and symmetrical, cases with unilateral cerebellar abnormalities are reported. [9] The abnormal cerebellar enhancement in our case appeared linear and may represent leptomeningeal or intravascular enhancement. The abnormal enhancement in this condition may reflect leptomeningeal inflammation, encephalitis, or perivenular compromise of the blood-brain barrier with alterations in cerebellar blood flow. [16] Differential diagnosis of postinfectious cerebellitis should include drug overdose. A history of recent exposure to drugs such as phenytoin, carbamazepine or alcohol must be ruled out in ever patient. [16] The sensitivity of MRI for the detection of cerebellitis is not known. A few patients with cerebellitis have presented with a normal MRI. [1-4] Abnormal noncontrast MRI findings in cerebellitis have only been described in a few case reports, many of which occurred in young children. [5-11] Isolated cerebellar abnormalities were noted, including parenchymal hyperintensities on T2-WI, swelling, and secondary obstructive hydrocephalus. Follow-up studies showed reversals of the acute changes and the development of atrophy years later in severe cases [5]. Abnormal MRI enhancement may be seen in some [1,2] but not all cases [3,7,9,11] in acute and subacute stages of the disease. SUMMARY SUMMARY Benign regressive postinfectious neurological disorders (BRPIND) comprises a group of poorly understood inflammatory/demyelinating white matter disorders of cerebrum, cerebellum and spinal cord. that is characteristically postinfectious in nature. It is unclear what are the triggers and effector mechanisms resulting in white matter insult, though tantalizing clues have emerged. These disorders exist on a continuum of postinfectious
- 12. neuroinflammatory and white matter demyelinative background that includes Guillain-Barre syndrome (GBS), acute disseminated encephalomyelitis (ADEM), Neuromyelitis Optica (NMO), optic neuritis, transverse myelitis and postinfectious cerebellitis. Each of these disorders differs in the spatial and temporal restriction of inflammation within the nervous system. However, clinical and pathologic studies support the notion that there are many common features of the inflammation and white matter demyelination that is postinfectious or postvaccinal in nature. [16] These disorders might coexist in various combinations in the same patient or might present clinically as an isolated disease. It looks like that the division between these postinfectious disorders is indistinct, which is suggestive of a clinical continuum. These disorders simply represent a single disease with different clinical presentations. Myelin basic protein (which is the main antigen that is targeted in the immune mechanism that end in myelin destruction) is different in different parts of the CNS. The myelin basic protein in the peripheral nerves is different from that of the CNS and this might explain why the demyelinative process may preferentially involves some parts of the CNS and spare other parts in different patients (depending upon the antigenic properties of the myelin basic protein of the involved sites) resulting in a protean clinical presentations of the same disease in different patients. Different areas of the white matter within the CNS and the peripheral nervous system are targeted by the inflammatory demyelinating pathological process in various combinations in different patients depending upon the antigenic properties of the myelin basic protein in these areas resulting in some patients having their optic nerves, cerebrum, and spinal cord involved (acute disseminated encephalomyelitis), other patients having their optic nerves and spinal cord involved (neuromyelitis optica) and so on. [27] Myelin destruction and inflammatory white matter demyelination is an immune-mediated mechanism in Benign regressive postinfectious neurological disorders (BRPIND) that is triggered by antecedent infection. The immune mechanisms include antibody-mediated complement dependant myelinolysis, T-cell mediated lysis of Schwann cells and T -cell mediated induction of an immune reaction with release of cytokines and recruitment of inflammatory cells including macrophages. The description of these immune mediated mechanisms are beyond the scope of this case record. Readers are referred to references 22-27 Postinfectious cerebellitis may be detected by MRI in adults and may include abnormal contrast enhancement. The changes on noncontrast MRI are similar to those noted in young children with cerebellitis and most likely reflect the reversible, inflammatory nature of the syndrome. The prognosis of acute cerebellitis is usually good. Even patients with severe symptoms and increased intracranial pressure can recover completely without any sequelae. Steroids are the first line of treatment when symptoms are moderate to severe; however, most patients will recover without steroids or any specific treatment [13, 14]. Sudden deaths have been reported following fulminant cerebellitis [15]. Death in acute cerebellitis is usually due to severe cerebellar swelling resulting in transtentorial and transforaminal herniations. [11,12] We conclude that MRI is an important tool in the diagnosis of acute cerebellitis, especially for cases in which the symptoms are mild and cerebrospinal fluid is normal. Although the cerebellum may show contrast enhancement, MRI findings of acute cerebellitis are very specific and There is no need for contrast enhancement once the characteristic cerebellar C-shaped MRI T2 white matter hyperintensities are demonstrated. [16] Addendum A new version of this PDF file (with a new case) is uploaded in my web site every week (every Saturday and remains available till Friday.) To download the current version follow the link quot;http://pdf.yassermetwally.com/case.pdfquot;. You can also download the current version from my web site at quot;http://yassermetwally.comquot;. To download the software version of the publication (crow.exe) follow the link:
- 13. http://neurology.yassermetwally.com/crow.zip The case is also presented as a short case in PDF format, to download the short case follow the link: http://pdf.yassermetwally.com/short.pdf At the end of each year, all the publications are compiled on a single CD-ROM, please contact the author to know more details. Screen resolution is better set at 1024*768 pixel screen area for optimum display. For an archive of the previously reported cases go to www.yassermetwally.net, then under pages in the right panel, scroll down and click on the text entry quot;downloadable case records in PDF formatquot; REFERENCES References 1. Bakshi R, Kinkel PR, Mechtler LL, Bates VE, Kinkel WR. Magnetic resonance imaging findings in acute cerebellitis. Clin Imaging. 1998;22:79-85. 2. Ravi V, Rozen T. Acute cerebellitis: MRI findings. Neurology. 2000;54:213. 3. Case records of the Massachusetts General Hospital: case 38-1996. N Engl J Med. 1996;335:1829-1834. 4. Mario-Ubaldo M. Cerebellitis associated with Lyme disease. Lancet. 1995;345:1060. 5. Hayakawa H, Katoh T. Severe cerebellar atrophy following acute cerebellitis. Pediatr Neurol.1995;12:159- 161. 6. Hayashi T, Ichiyama T, Kobayashi K. A case of acute cerebellar ataxia with an MRI abnormality. Brain Dev. 1989;11:435-436. 7. Horowitz MB, Pang D, Hirsch W. Acute cerebellitis: case report and review. Pediatr Neurosurg. 1991- 92;17:142-145. 8. Hurst DL, Mehta S. Acute cerebellar swelling in varicella encephalitis. Pediatr Neurol. 1988;4:122-123. 9. Lester A, Alpigiani MG, Franzone G, Cohen A, Puleo MG, Tortori-Donati P. Magnetic resonance imaging in right hemisphere cerebellitis associated with homolateral hemiparesis. Childs Nerv Syst. 1995;11:118-120. 10. Nakagawa E, Yamanouchi H, Sakuragawa N, Takashima S. Vermis lesions in acute cerebellar ataxia: a sequential imaging study. Brain Dev.1994;16:488-490. 11. Shoji H, Hirai S, Ishikawa K, et al. CT and MR imaging of acute cerebellar ataxia. Neuroradiology. 1991;33:360-361. 12. Tlili-Graiess K, Mhiri Souei M, Mlaiki B, Arifa N, Moulahi H, Jemni Gharbi H, et al. Imaging of acute cerebellitis in children. Report of 4 cases. J Neuroradiol 2006;33: 38–44. 13. De Bruecker Y, Claus F, Demaerel P, Ballaux F, Sciot R, Lagae L, et al. MRI findings in acute cerebellitis. Eur Radiol 2004;14:1478–83. 14. Aylett SE, O’Neill KS, De Sousa C, Bitton J. Cerebellitis presenting as acute hydrocephalus. Child Nerv Syst 1998;14:139–41.
- 14. 15. Levy EI, Harris AE, Omalu BI, Hamilton RL, Branstetter BF 4th, Pollack IF. Sudden death from fulminant acute cerebellitis. Pediatr Neurosurg 2001;35:24–8. 16. Metwally, MYM: Textbook of neuroimaging, A CD-ROM publication, (Metwally, MYM editor) WEB-CD agency for electronic publication, version 9.2a April 2008 17. Miller HG, Stanton JB, Gibbons JL. Para-infectious encephalomyelitis and related syndromes. J Med 1956;NS XXV(100):427-505. 18. Scott TFM. Postinfectious and vaccinal encephalitis. Med Clin North Am 1967;51:701-16. 19. Lisak RP, Behan PO, Zweiman B, Shetty T. Cell-mediated immunity to myelin basic protein in acute disseminated encephalomyelitis. Neurology 1974;24:560-4. 20. Johnson RT, Griffin DE, Hirsch RL, et al. Measles encephalomyelitis clinical and immunologic studies. N Engl J Med 1984;310:137-41. 21. Johnson RT. The pathogenesis of acute viral encephalitis and postinfectious encephalomyelitis. J Infect Dis 1987;155:359-64. 22. Phol-Koppe A, Burchett SK, Thiele EA, Hafler DA. Myelin basic protein reactive Th2 T cells are found in acute disseminated encephalomyelitis. J Neuroimmunol 1998;91:19-27. 23. Ichiyama T, Shoji H, Kato M, et al. Cerebrospinal fluid levels of cytokines and soluble turnout necrosis factor receptor in acute disseminated encephalomyelitis. Eur J Pediatt 2002;161:133-7. 24. Hemmer B, Cepok S, Nessler S, Sommer N. Pathogenesis of multiple sclerosis: an update on immunology. Curr Opin Neurol 2002; 15:227 31. 25. Stocks M. Genetics of childhood disorders: XXIX. autoimmune disorders, part 2: molecular mimicry. J Am Acad Child Adolesc Psychiatry 2001;40:977-80. 26. Khong PL, Ho HK, Cheng PW, Wong VCN, Gob W, Chan FL. Childhood acute disseminated encephalomyelitis: the role of brain and spinal cord MRI. Pediatr Radiol 2002;32:59-66. 27. Hughes, RA. Inflammatory neuropathies. Bailliere's Clinical neurology Vol. 3, No 1, April 1994 45-72 28. Dale RC, Church AJ, Cardoso F, Goddard E, Cox TC, Chong WK, et al. Post streptococcal acute disseminated encephalomyelitis with basal ganglia involvement and auto-reactive antibasal ganglia antibodies. Ann Neurol 2001;50:588-95. 29. Idrissova ZR, Boldyreva MN, Dekonenko EP, Malishev NA, Leontyeva IY, Martinenko IN, et al. Acute disseminated encephalomyelitis in children: Clinical features and HLA-DR linkage. Eur J Neurol 2003;10:537-46. 30. Kumar A, Swamy HS, Santhosh V, Taly AB, Arunodaya GR, Shankar SK. Pathology of allergic encephalomyelitis following Semple type antirabies vaccine from India. Neurol Infect Epidemiol 1997;2:239- 48.