Hemoglobin final
•Download as PPTX, PDF•
2 likes•632 views

Hemoglobin , derivatives and Structural / synthesis abnormalities

Recommended
More Related Content
What's hot
What's hot (20)
Similar to Hemoglobin final
Similar to Hemoglobin final (20)
Recently uploaded
Recently uploaded (20)
Hemoglobin final
- 2. WAY TO…… RBC – in 1665 by Marcello Malpighi Isolation of Hb – in 1862 by Felix Hoppe Role of Hb for O2 transport – in 1904 by Christian Bohr Structure of Hb – in 1912 by Kuster Synthesized Hb in lab – in 1920 by Hans Fischer Three Dimensional stru. Of Hb – in 1962 by Perutz 4/10/2021 Dr. V.P.Shah
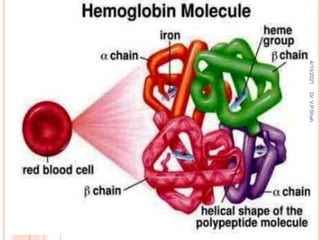
- 4. HEMOGLOBIN Normal Level in blood: Male : 14-16g/dl Female: 13-15g/dl HbA , HbA2, HbF HbA : 2 alpha and 2 beta chain – mole. Weight – 67,000 daltons (Normal level – 97%) HbF : 2 alpha and 2 gamma chains (normal level 1%) HbA2: 2 alpha and 2 delta chain (Normal level – 2%) 4/10/2021 Dr. V.P.Shah
- 5. HEMOGLOBIN = HEME + GLOBIN Heme: Derivatives of Porphyrins Porphyrins are cyclic compounds formed by fusion of 04 pyrrole ring linked by methenyl bridges Presence of Iron in Heme – Ferroprotoporphyrins Pyrrole rings are named as I,II, III, IV Bridges are named as alpha, Beta, Gamma & Delta Possible areas of substitution are 1-8 Propionyl, Acetyl, Methyl and Vinyl The two hydrogen atoms in the –NH groups of pyrrole rings (II and IV) are replaced by ferrous iron (Fe++) Occupy the centre of the compound ring structure Establish linkages with all the four nitrogens of all the pyrrole rings. 4/10/2021 Dr. V.P.Shah
- 7. STRUCTURE OF HEMOGLOBIN Heme is located in a hydrophobic cleft of Globin Chain 04 Heme Residue per Hb Molecule Iron – Central Position of Porphyrin Ring – Linked with pyrrole N by 04 cordinate valency bond Reduced state – Fe++ -- Ferous Ion – 06 valency (5th with imidazole N of Proximal Histidine) & (6th valency Iron bind with O2 and formed Hydrogen Bond with Imidazole N of Distal Histidine) 4/10/2021 Dr. V.P.Shah
- 8. STRUCTURAL ORGANIZATION OF HB Primary structure: Globin subunit Secondary structure: Each globin chain contains several helical segments separated by short non-helical segments 8 helical segment in β,γ & δ chain designed as A, B, C, D, E, F, G & H α chain lacks D segment & contains 7 helices 4/10/2021 Dr. V.P.Shah
- 11. Tertiary structure: Globin chain are highly compact structure Hydrophilic residues directed to exterior & hydrophobic residues directed to interior Heme pocket Interaction of heme with globin by non-covalent binding Quaternary structure: Tetramer of 4 polypeptide chain & 4 heme are held together in definite arrangement or conformation by non- covalent bonds such as hydrogen bond, ionic interaction, hydrophobic interactions and van-der waals forces to form quaternary structure. 4/10/2021 Dr. V.P.Shah
- 14. Hb A1 Tetramer of 2 α subunit & 2 β subunit (α2β2) 90-95 % major form of Hb Affinity of Hb A1 for O2 is lower than Hb F because of high affinity of Hb A1 for 2,3 BPG Hb A2 Tetramer of 2 α subunit & 2 δ subunit (α2δ2) Minor form 2% 4/10/2021 Dr. V.P.Shah
- 15. FETAL HEMOGLOBIN (HBF) HbF has 2 Alpha chains and 2 Gamma Chains (having 146 Amino Acid) Increased Solubility of deoxy HbF Slower Electrophoretic Mobility of HbF Increased resistance of HbF to alkali denaturation HbF has decreased interaction with 2,3 BPG ODC of Fetus and Newborn are shifted to left. It helps in facilitating transplacental oxygen transport. HbF synthesis starts by 7th Week of gestation At birth 80% of Hb is HbF During first 06 months of life, it decreases to about 5% of total 4/10/2021 Dr. V.P.Shah
- 16. WHY HB F HAS HIGH AFFINITY FOR O2 THAN HB A In γ chain one of the basic AA His-143 replaced by neutral Serine AA This removes +ve charge from 2,3 BPG binding site & reduces the affinity of fetal Hb for 2,3 BPG Thus ↑ ing the O2 binding affinity of fetal Hb relative to that of maternal Hb. 4/10/2021 Dr. V.P.Shah
- 18. Globin synthesis, starts at 3rd week of gestation Embryonic Haemoglobin Gower I ( z2e2) Haemoglobin Portland ( z2g2) Haemoglobin Gower II (a2e2) Fetal : HbF (a2g2), HbA (a2b2) Adult : HbA, HbA2 ( a2d2), HbF. SYNTHESIS OF GLOBIN 4/10/2021 Dr. V.P.Shah
- 21. GLYCOSYLATED HB (HBA1C) Aldehyde group of Glucose react with amino group of N-terminal residue (Valine) of β chain in a non-enzymatic reaction 4 to 6 % of total Hb in blood Increased in DM Estimation of Glycosylated Hb useful for monitoring long term control of hyperglycemia in DM patients 4/10/2021 Dr. V.P.Shah
- 23. TRANSPORT OF OXYGEN BY HEMOGLOBIN Ideal Respiratory Pigment: Transport Large Quantity of Oxygen Great Solubility Take up and release oxygen powerful buffer 4/10/2021 Dr. V.P.Shah
- 24. TRANSPORT OF O2 FROM LUNGS TO TISSUES Oxygenation of Hb accompanied by conformation changes in the tertiary & quaternary structure of hb Conformational changes in Hb: Widening of heme pockets Movement of subunits Movement of the iron atom Disruption of salt bridge 4/10/2021 Dr. V.P.Shah
- 26. OXYGEN DISSOCIATION CURVE It helps in understanding of Hemoglobin ability to load and unload oxygen at physiological pO2 Sigmoid Shape of ODC is due to allosteric effect or cooperativity Hill Equation expresses the equilibrium of Hb with oxygen. Hb carries 20 ml of O2/dl (Hb level – 15 g/dl) pO2 values in : Inspired air 158 mm Hg Alveolar Air 100 mm Hg Blood in Lungs 90mm Hg (97% saturated) Capillary Bed 40 mm Hg (Hb saturation :60%) 4/10/2021 Dr. V.P.Shah
- 28. FACTORS AFFECTING OXYGEN DISSOCIATION CURVE Heme-Heme Interaction and Cooperativity Positive Co operativity: Binding of Oxygen to one Heme Residue increases affinity of remaining heme residue for O2 (Homotropic Interaction) Heterotropic Interaction: Binding of 2,3-BPG lowers affinity for O2 Oxy Hb as R Form (Relaxed Form) De oxy Hb as T form (Tight Form) When Oxygenation occurs salt bonds are broken down subsequently Hb – HbO2 – HbO4-HbO6-HbO8 4/10/2021 Dr. V.P.Shah
- 31. THE BOHR EFFECT It is influence of pH and PCO2 to facilitate oxygenation of Hb in lungs and deoxygenation at the tissues In tissues: pCO2 elevated – pH falls – H+ concentration increases (formation of Metabolic acids.. Lactate) O2 affinity will decreases – ODC shifted to right- O2 released In Lungs: pCO2 low – pH high – pO2 significantly elevated – O2 binds with Hb – ODC shifted to left 4/10/2021 Dr. V.P.Shah
- 33. Effect of Temperature: - p50 means Hb is half saturated with O2 - Elevation of temp from 20 to 37 C causes 88% increase in p50 - Hypothermia – Metabolic Demand is low – shift of ODC to left – less O2 to the tissues - Febrile Condition – Increase needs of O2 – Shift of ODC to right 4/10/2021 Dr. V.P.Shah
- 34. THE CHLORIDE SHIFT – HAMBURGER EFFECT 4/10/2021 Dr. V.P.Shah
- 36. EFFECT OF 2,3-BPG Normal blood level- 15±1.5mg/g Hb Higher in Younger children It is intermediate of Glycolytic Pathway – Rapaport Leubering Cycle/Shunt It binds with Deoxy Hemoglobin and stabilize T form During oxygenation 2,3 BPG released HbF contains gamma chain – having inability to bind with 2,3 –BPG – So it has high affinity to O2 4/10/2021 Dr. V.P.Shah
- 40. Right shift (easy oxygen delivery) High 2,3-BPG High H+ High CO2 HbS Left shift (give up oxygen less readily) Low 2,3-BPG HbF HB-OXYGEN DISSOCIATION CURVE 4/10/2021 Dr. V.P.Shah
- 41. HEMOGLOBIN – AS BUFFERING AGENT With the pH range of 7.0 to 7.8, most of the physiological buffering action of Hb is due to the “imidazole” group of amino acid “histidine”. Imidazole contains two groups 1. Fe++ containing group which is concerned with carriage of O2, and 2. Imidazole N2 group, which can give up H+ (proton) and accept H+ depending on the pH of the medium. Thus, buffering capacity of Hb is due to the presence of “Imidazole” nitrogen group which remains dissociated in acidic medium and conjugate base forms. 4/10/2021 Dr. V.P.Shah
- 42. DERIVATIVES OF HEMOGLOBIN 1. Action of Acids & Alkalis on Hb 4/10/2021 Dr. V.P.Shah
- 43. DERIVATIVES OF HEMOGLOBIN 2. Haemin It is chemically haematin hydrochloride. Boiling oxy-Hb with NaCl and glacial acetic acid. Iron is oxidized in ferric form – bind with negatively charged Cl- - to form hematin chloride Laboratory – blood are heated with Nippe’s Fluid – forms dark brown rhombic crystals are seen Sensitive test – answered by heme part of blood of all species 3. Haemochromogen Haem and the ferrous porphyrin complexes react readily with basic substances such as hydrazines, primary amines, pyridines, or an imidazole such as the amino acid histidine, resulting compound is called a haemochromogen (haemochrome) 4/10/2021 Dr. V.P.Shah
- 44. 4. Haematoporphyrin Mixing Blood with Sulphonic Acid 5. Haematoidin Produced by the breakdown of Hb in the body. Found as yellowish-red crystals in the region of old blood extravasation. 6. Methaemoglobin 4/10/2021 Dr. V.P.Shah
- 45. MET HEMOGLOBIN Ferous ion of Hb is oxidized to Ferric state – Met – HB is formed ----- (Oxidized Stress- Generation of Free Radicals) Prevented by : Met Hb reductase Enzyme System – using NADH and Cytochrome b5 Others – NADPH dependent System & Glutathione Dependent Met – Hb reductase Normal level – less than 1% Congenital / Acquired Met hemoglobinemia Cyanosis 4/10/2021 Dr. V.P.Shah
- 46. METHEMOGLOBIN - CAUSES certain oxidant drugs or exposure to certain poisons, e.g. chlorates, acetanilid, nitrites, nitrobenzene, antipyrin, phenacetin, sulphonal, and perhaps most important, the sulphonamide drugs. Use of Potassium Ferricyanide Nitrobenzene is used in manufacture of shoe dyes,floor polishes, cosmetics and explosives. Fumes from carbon arcs contain nitrous oxide Familial methaemoglobinaemia An inherited disorder due to lack or absence of the enzyme methaemoglobin reductase, 4/10/2021 Dr. V.P.Shah
- 47. MET – HB - SYMPTOMS 4/10/2021 Dr. V.P.Shah
- 48. CONGENITAL MET HEMOGLOBINEMA Cytochrome b5 reductase deficiency Cynosis at birth Blood level – 10 -15% Treatment – Administration of Methylene Blue 100- 300 mg/day, Ascorbic Acid 4/10/2021 Dr. V.P.Shah
- 49. MET-HB : TREATMENT Injection of intravenous glucose or methylene blue, which helps to reduce Met-Hb (Fe+++) to Hb (Fe++), Methylene blue activates NADH or NADPH dependant methaemoglobin reductase/ diaphorase I. Administration of ascorbic acid also helps in reduction. 4/10/2021 Dr. V.P.Shah
- 50. CARBOXY HEMOGLOBIN Hb binds with CO 200 times more affinity for Hb Not suitable for O2 transport One mole. Bind with one monomer of Hb – increase affinity of other monomer for o2 O2 bind with this monomer can not release Decrease availability of O2 to tissues 4/10/2021 Dr. V.P.Shah
- 51. CARBOXY HB CO – Colorless, Odorless, tasteless gas – generated by incomplete combustion Specifically CO Poisoning is seen in Mine Workers & Automobile Industries Normal Blood level – 0.16% Clinical Manifestation: Level exceeds 20% Symptoms – Breathlessness, Nausea, Headache, Vomiting, Chest pain Treatment – O2 , Hyperbaric Oxygen 4/10/2021 Dr. V.P.Shah
- 52. CARRIAGE AS CARBAMINO HEMOGLOBIN - 15% CO2 is carried as Carbamino Hemoglobin - CO2 bind with Hb as a Carbamino Complex - R-NH2 + CO2 --- R-NH-COOH 4/10/2021 Dr. V.P.Shah
- 53. SULF HEMOGLOBINEMIA Reactions of Oxy Hb with Hydrogen sulfide Causes: Sulfonamide, Phenacetin, acetanilide, dapsone Causes Basophilic stippling of RBC Nitric Oxide NO – Hb bind with high affinity Increase half life of NO T – state bind with NO Delivers NO to tissue capillaries 4/10/2021 Dr. V.P.Shah
- 54. HCN & CYANIDE Cyanides resides -inhibits cytochrome oxidase a3 of electron transport chain and stops cellular respiration. Treatment --- Formation of Cyanmethemoglobin – Not Toxic – Injection of Sodium Nitrite – Methemoglobin – Binds with Cyanide – Cyanmethemoglobin – slowly convert into Cyanate and Hb Use of Sodium Thiosulphate - Thiocyanate 4/10/2021 Dr. V.P.Shah
- 55. What is the importance of Spectroscopy in Clinical Chemistry and Forensic Science? Methaemoglobin is not found in blood normally. Why? Oral Administration of Methylene Blue Dye or Ascorbic Acid reverses cyanosis in Methaemoglobinemias. Why? Why Coal Mine workers are at risk at Carbon Monoxide Poisoning? 4/10/2021 Dr. V.P.Shah
- 56. HAEMOGLOBINOPATHIES Inherited disorders of hemoglobin synthesis (thalassaemias) or structure (sickle cell disorders) that are responsible for significant morbidity and mortality These disorders result in errors in oxygen-carrying capacity of hemoglobin Diseases linked to genetic predisposition Sickle cell and Thalassaemia are inherited disorders that are passed on from parents to children through unusual haemoglobin genes 4/10/2021 Dr. V.P.Shah
- 58. Presentation -: Disease: they inherit two unusual haemoglobin genes – one from their mother, and one from their father. Trait/Carrier: People who inherit just one unusual gene are known as ‘carriers’. (‘trait’.) Carriers are healthy and do not have the disorders 4/10/2021 Dr. V.P.Shah
- 61. Quantitative Hemoglobinopathies: lack or ↓ ed synthesis of either α or β globin chain, altered combination of normal chain e.g. Thalassemia Qualitative /Structural Hemoglobinopathies: altered sequence of AAs, usually in one of the constituent chains e.g. Sickle cell disease, Hb C d’se, Hb M d’se 4/10/2021 Dr. V.P.Shah
- 62. HEMOGLOBIN VARIANT CLASSIFICATION: Sickle Syndromes Sickle cell trait (AS) Sickle Cell Disease with SS, SC , SD, SO, varieties and S Beta Thalassemia Unstable Hemoglobin:Congenital Heinz body Anemia, Hb Zurich, Hb Kohn Hemoglobin with abnormal oxygen affinity High Affinity : Polycythemia (Familial) , Hb Chesapeake, Hb Kansas Low Affinity : Cyanosis (Familial) , HbM, Hb Rainier Structural Variations leading to Thalassemia Alpha Thalassemia : Hb Constant Spring, Delta beta thalassemia, Hb Lepore Beta Thalassemia : Hb Quong sze 4/10/2021 Dr. V.P.Shah
- 63. Sickle cell Hb (Hb S): Abnormal Hb causes sickle cell anemia depending on mode of inheritance Sickle cell anemia: results from homozygous inheritance of Hb S gene (Hb SS) Homozygous : Contains 0 - 20% HbA and 80 -100% HbS Sickle cell trait: refers to the heterozygous inheritance of Hb S gene (Hb AS) Heterozygous: Contains 60 -80% HbA and 20 -40% HbS 4/10/2021 Dr. V.P.Shah
- 64. GENETICS Hb S caused by substitution of a Valine for Glutamic acid residue as the 6th AA in β globin chain Results from a point mutation in DNA due to substitution of Thymine for Adenine Population affected: Africa, India, Middle East & Southern Europe 4/10/2021 Dr. V.P.Shah
- 65. Hb – S : Defect : α 2 β 2 A6Val(Glutamic Acid) – Suggest replacement of Gluctamic Acid(2 carboxy polar residue) by Valine(Non polar residue) Its Effect: Alters distribution of +ve & -ve charges on protein surface. Difference between HbA and HbS As HbA, HbS also contains Oxygenated and Deoxygenated forms while in circulation. But Deoxygenated form of HbS is only 2 % soluble then Deoxygenated HbA and 1 % soluble then own oxy form. 4/10/2021 Dr. V.P.Shah
- 66. PATHOPHYSIOLOGY The biochemical basis for the structural alterations of Hb S Formation of rigid erythrocytes Obstruction of small blood vessels by rigid erythrocytes (sickle cell crisis) Hemolytic anemia 4/10/2021 Dr. V.P.Shah
- 67. PATHOPHYSIOLOGY β α α β β α α β β α α β β α α β Complimentary Site Sticky Patch OXYGENATED DEOXYGENATED 4/10/2021 Dr. V.P.Shah
- 69. During Deoxygenated state of HbS, Sticky patch bind to complimentary site on another Deoxygenated HbS molecule and causes polymerization of Deoxy HbS which forms long fibrous precipitates extends throught Red Blood Cells – Causes distortion and lysis of RB Cell 4/10/2021 Dr. V.P.Shah
- 71. CLINICAL FEATURES Chronic hemolytic anemia Vaso-occlusive crisis Sickle cell trait show increased resistance to malaria specifically for plasmodium malaria 4/10/2021 Dr. V.P.Shah
- 73. FACTORS AFFECTING SEVERITY OF SICKLING ↓ O2 tension caused by high altitude ↑ CO2 concentration ↓ pH ↑ concentration of 2,3 BPG in RBC Hypoxia, Infections, Dehydration, Cold & acidosis 4/10/2021 Dr. V.P.Shah
- 74. DIAGNOSIS Microcytic Hypochromic anemia: Hb ↓ (6-8 gm/dl) ↑ Reticulocyte count Sickling of red cell seen in microscope Sickle cell solubility test: reducing agent such as sodium dithionate/ sodium metabisulfite Hb electrophoresis presence of Hb S Molecular Diagnosis: RFLP 4/10/2021 Dr. V.P.Shah
- 76. MANAGEMENT Symptomatic Mx Hydroxyurea: antisickling drug interfere with normal erythropoiesis in such a way to ↑ production of γ chains leading to ↑ synthesis of Hb F (interfere with sickling) Bone marrow transplantation 4/10/2021 Dr. V.P.Shah
- 77. The efficacy of hydroxyurea in the treatment of sickle cell disease is generally attributed to its ability to boost the levels of fetal hemoglobin (Hb F,α2γ2). This lowers the concentration of Hb S within a cell resulting in less polymerization of the abnormal hemoglobin. 4/10/2021 Dr. V.P.Shah
- 78. MAY BE DUE TO……… HYDROXYUREA is cytotoxic to the more rapidly dividing late erythroid precursors, an effect that leads to the recruitment of early erythroid precursors with an increased capacity to produce Hb F. interrupt the transcription factors that selectively bind to promoter or enhancer regions around the globin genes, thereby altering the ratio of Hb A to Hb F nitric oxide-derived mechanism for Hb F induction increases Hb F production by inhibiting ribonucleotide. 4/10/2021 Dr. V.P.Shah
- 79. Imp: Resistant to Malaria in patients with Sickle cell ds. - Malaria parasite spends a part of its life cycle in erythrocyte – In HbS interrupts the parasite cycle - MP increases the acidity – increase sickling of Erythrocytes to 40% - Concentration of K is low in sickled cells – unfavorable for MP to survive. Increase incidence of Salmonella 4/10/2021 Dr. V.P.Shah
- 81. Group of genetically transmitted disorders of Hb synthesis characterized by Impairment / Imbalance in syn. Of Globin chain Thalasa means sea Classified based on globin chain or inheritance patterns 4/10/2021 Dr. V.P.Shah
- 83. BETA THALASSEMIA Structurally normal but defective synthesis/production of beta globin chain of Hb Types: major & minor Excess of alpha chain to form α4 precipitate rapidly within the RBC as heinz bodies, cell rupture & death ↑ con. of Hb F & Hb A2 4/10/2021 Dr. V.P.Shah
- 84. CAUSES Mutation of β globin chain (frame shift/ non-sense mutations) Defective transcription due to mutation in promoter or enhancer region Defective RNA processing due to defective splicing Defective translation due to base exchange mutation Frame shift mutation 4/10/2021 Dr. V.P.Shah
- 85. THALASSEMIA MAJOR Genetics: absence of both β globin genes, homozygous state Age of onset & sex: early life (6th month of age), affect both sex equally Geographical prevalence: mediterranean region like south africa, india& south east asia 2-15% incidence 4/10/2021 Dr. V.P.Shah
- 86. CLINICAL FEATURES Hemolytic anemia (cooley’s anemia) Splenomegaly Hepatomegaly 4/10/2021 Dr. V.P.Shah
- 87. COOLEY’S ANEMIA Severe anemia of Infancy or early childhood Features – Mongoloid Features having stunted growth, pallor, Icterus, Splenomegaly Hypochromic Microcytic Anemia Blood Smear – Basophilic Strippling (a blood smear in which erythrocytes display small dots at the periphery. These dots are the visualization of ribosomes , Target Cell ++ 4/10/2021 Dr. V.P.Shah
- 88. Skull Bone – Hair on end experience – Radiographic appearance on a skull which results from a periosteal reaction manifesting as perpendicular trabeculations interspersed radiolucent marrow hyperplasia 4/10/2021 Dr. V.P.Shah
- 89. DIAGNOSIS Microcytic hypochromic anemia ↓ Hb level (<7gm/dl) Reticulocytosis Fragmentation of erythrocytes Presence of inclusion bodies Serum iron ↑ Serum ferritin ↑ 4/10/2021 Dr. V.P.Shah
- 90. Hb electrophoresis: absence of Hb A (α2β2) ↑ Hb A2 (α2δ2) ↑ Hb F (α2γ2) Hair on end appearance in skull in X-ray Prenatal diag. chorionic or amniotic fluid sampling 4/10/2021 Dr. V.P.Shah
- 91. TREATMENT Blood transfusion Folic acid supplements Iron chelation therapy (Desferrioxamine) Bone marrow transplantation (useful in young HLA matched siblings) 4/10/2021 Dr. V.P.Shah
- 92. THALASSEMIA MINOR Genetics: caused by the absence of only one globin chain of Hb & heterozygous Benign condition, mild anemia Hb A +nt Hb A2 ↑ 4/10/2021 Dr. V.P.Shah
- 93. THALASSEMIA INTERMEDIA Caused by either combination of homozygous mild β thalassemia & α thalassemia Β thalassemia & hereditary persistance of Hb or Hb lepore (caused by defect DNA recombination of δ & β chain of Hb) Moderate degree anemia, splenomegaly & gall stone ↑ Hb F 4/10/2021 Dr. V.P.Shah
- 94. Α-THALASSEMIA Are disorder of Hb synthesis caused by defective syn. of α globin chain Resulting excess β & γ globin chain ↓ Hb A1, Hb A2, Hb F Causes: point mutation, insertion & deletion 4/10/2021 Dr. V.P.Shah
- 96. Silent thalassemia: one globin chain absent,no clinical effect α thalassemia trait: two globin chain absent, benign condition α thalassemia minor (Hb H d’se): three globin chain absent, β4 accumulation called Hb H, mild anemia α thalassemia major (Hb Bart’s):four globin chain absent, γ4 accumulation called Hb bart, also called hydrops fetalis 4/10/2021 Dr. V.P.Shah
- 97. HB C Replacement of 6 position of beta chain Glutamic Acid by lysine More common on black race AC heterozygotes do not show any clinical manifestations 4/10/2021 Dr. V.P.Shah
- 98. HB E It is second most common hemoglobin variant Replacement of beta 26 glutamate by lysine More prevalent in west Bengals in India 4/10/2021 Dr. V.P.Shah
- 99. HB D Replacement of beta 121 glutamate by glutamine (HbD Punjab) HbSD disease is a severe condition 4/10/2021 Dr. V.P.Shah
- 100. HB SABINE Defect : α2 A β2 91 Proline Substitution of β chain of Proline for leucine at 91, this makes possible for formation of methaemoglobin. Globin moiety not attached to Heme gets precipitated in the erythrocyte – Inclusion Bodies: attached to Cell Membrane – Osmotic Damage causing hemolysis. 4/10/2021 Dr. V.P.Shah
- 101. HB CHESAPEAKE Defect: α292Arg(Lysine) β2 A High affinity to oxygen – decreased release to tissue causing tissue hypoxia – compensatory Polycythemia 4/10/2021 Dr. V.P.Shah
- 102. HB RAINIER Tyrosine replaced by Histidine at 145causing stabilize R form and increase O2 Affinity Abnormal Hb which interferes with m –RNA formation (Structural Variants) 4/10/2021 Dr. V.P.Shah
- 103. HB CONSTANT SPRING Produce unstable m –RNA, UAA stop codon has mutated CAA which codes for Glutamine hence α chain are 31 residues longer than usual. The longer m –RNA (i.e 172 a.a) is unstable and gets degraded readily. 4/10/2021 Dr. V.P.Shah
- 104. QUESTIONS Describe the structure & function of Hb Describe the disorder of Hb Describe the causes, features & diagnosis of sickle cell disease Describe the causes, features & diagnosis of beta thalassemia Mention five disorders characterized by altered Hb structure & function. Mention the underlying defect & major clinical features of each condition 4/10/2021 Dr. V.P.Shah
- 105. QUESTIONS What are causes of MetHb? Mention the effect of MetHb on oxygenation of Hb. How it is detected. List of two compounds used in treatment Mention different types of Hb their composition & significance Describe ODC of Hb & mention the factors causing shift of ODC Describe changes occuring during CO2 transport 4/10/2021 Dr. V.P.Shah
- 106. QUESTIONS What are tactoid cells how they are formed mention their significance Describe different form of alpha thal. & defect in each type. Abnormal Hb Role of 2,3 BPG in transport of O2 by Hb MetHb Give differentiation point for T & R form of Hb Hb S 4/10/2021 Dr. V.P.Shah
- 107. QUESTIONS Conformational changes occur during formation of oxy Hb What is Bohr effect Why Hb F high affinity for O2 than Hb A What is Glyco-Hb & its significance Why does person with sickle cell anemia show increased resistance to malaria Give factors affecting severity of sickling What is Hb M 4/10/2021 Dr. V.P.Shah
Editor's Notes
- Inclusion bodies in Erythrocytes[edit] Normally a red blood cell does not contain inclusions in the cytoplasm. However, it may be seen because of certain hematologic disorders. There are three kinds of erythrocyte inclusions: Developmental Organelles Howell-Jolly bodies: small, round fragments of the nucleus resulting from karyorrhexis or nuclear disintegration of the late reticulocyte and stain reddish-blue with Wright stain. Basophilic stipplings - these stipplings are either fine or coarse, deep blue to purple staining inclusion that appears in erythrocytes on a dried Wright stain. Pappenheimer bodies - are siderotic granules which are small, irregular, dark-staining granules that appear near the periphery of a young erythrocyte in a Wright stain. Polychromatophilic red cells - young red cells that no longer have nucleus but still contain some RNA. Cabot Rings - ring-like structure and may appear in erythrocytes in megaloblastic anemia or in severe anemias, lead poisoning, and in dyserythropoiesis, in which erythrocytes are destroyed before being released from the bone marrow. Abnormal Hemoglobin Precipitation Heinz bodies - round bodies, refractile inclusions not visible on a Wright stain film. It is best identified by supravital staining with basic dyes. Hemoglobin H Inclusions - alpha thalassemia, greenish-blue inclusion bodies appear in many erythrocytes after four drops of blood is incubated with 0.5mL of Brilliant cresyl blue for 20 minutes at 37 °C.