Downloaded 1,979 times









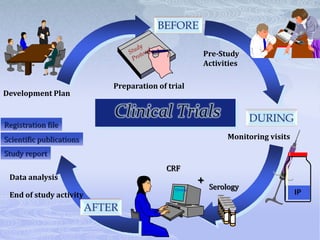




































































This document discusses the clinical trials process from protocol development through study completion. It covers developing the protocol, regulatory documents, patient recruitment, safety reporting, interim reports, and end of study activities. Key aspects include writing an approvable protocol, establishing an investigator site file, screening and enrolling suitable patients, maintaining safety oversight, and conducting closeout procedures. The goal is to provide guidance on managing all stages of a clinical trial.

















![roles and responsibilities of Investigator[663]](https://cdn.slidesharecdn.com/ss_thumbnails/investigator663-210616055819-thumbnail.jpg?width=640&height=640&fit=bounds)
























































