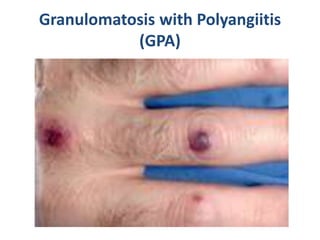
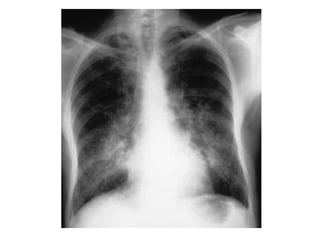
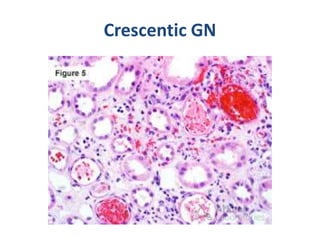
Vasculitis is characterized by inflammation of blood vessels. It can be primary or secondary to other diseases. Primary vasculitis syndromes include Granulomatosis with Polyangiitis (GPA, also known as Wegener's), which involves necrotizing granulomatous inflammation and vasculitis that commonly affects the lungs and kidneys. Diagnosis of GPA involves laboratory tests, biopsy of affected tissues showing vasculitis and granulomas, and testing positive for antineutrophil cytoplasmic antibodies (ANCA). Treatment is with corticosteroids and immunosuppressants to induce remission.




















![Symptoms and Signs
• Onset of granulomatosis with polyangiitis may be insidious or
acute; the full spectrum of the disease may take years to evolve.
• Some patients present initially with upper and lower respiratory
tract symptoms; at some point later, the kidneys are affected.
• In other patients, onset of systemic manifestations is relatively
acute; several organs and systems, such as the upper respiratory
tract, peripheral nervous system (causing multiple
mononeuropathy [mononeuritis multiplex]), kidneys (causing
glomerulonephritis), and lower respiratory tract (causing
hemorrhage,
• lung nodules, cavities, or a combination), are simultaneously
affected.](https://image.slidesharecdn.com/vasculitis-230326055300-8e4d2368/85/Vasculitis-pptx-23-320.jpg)