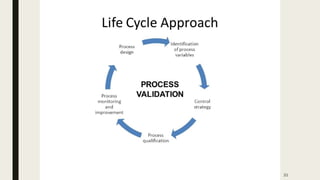
The document outlines US FDA guidelines on process validation for pharmaceuticals, emphasizing the integration of process validation activities within the product lifecycle. It details the three stages of process validation: process design, process qualification, and continued process verification, highlighting the regulatory requirements and the importance of documentation and risk-based decision making. The guidelines ensure that manufacturing processes consistently meet quality standards, ultimately protecting public health.