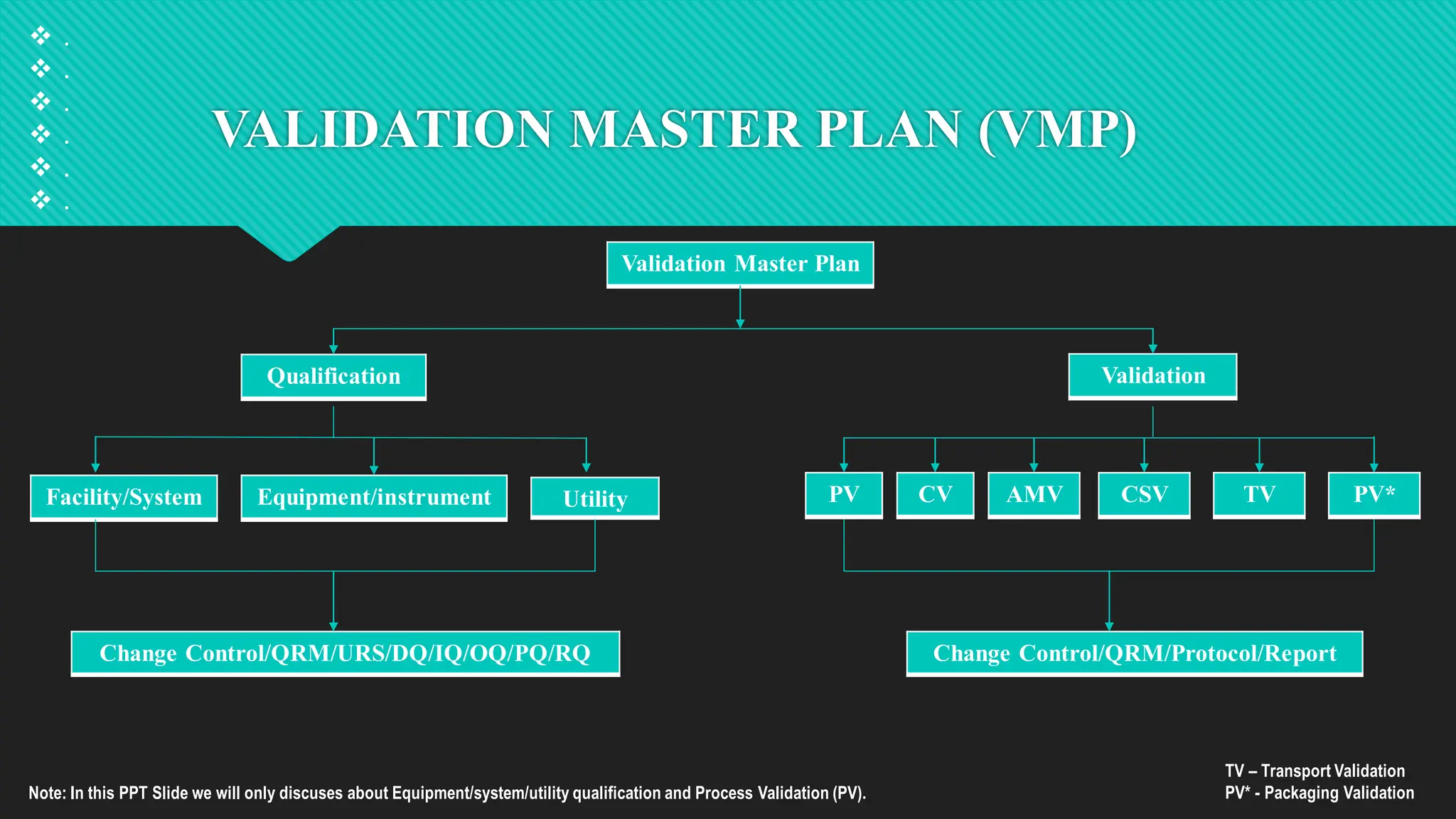
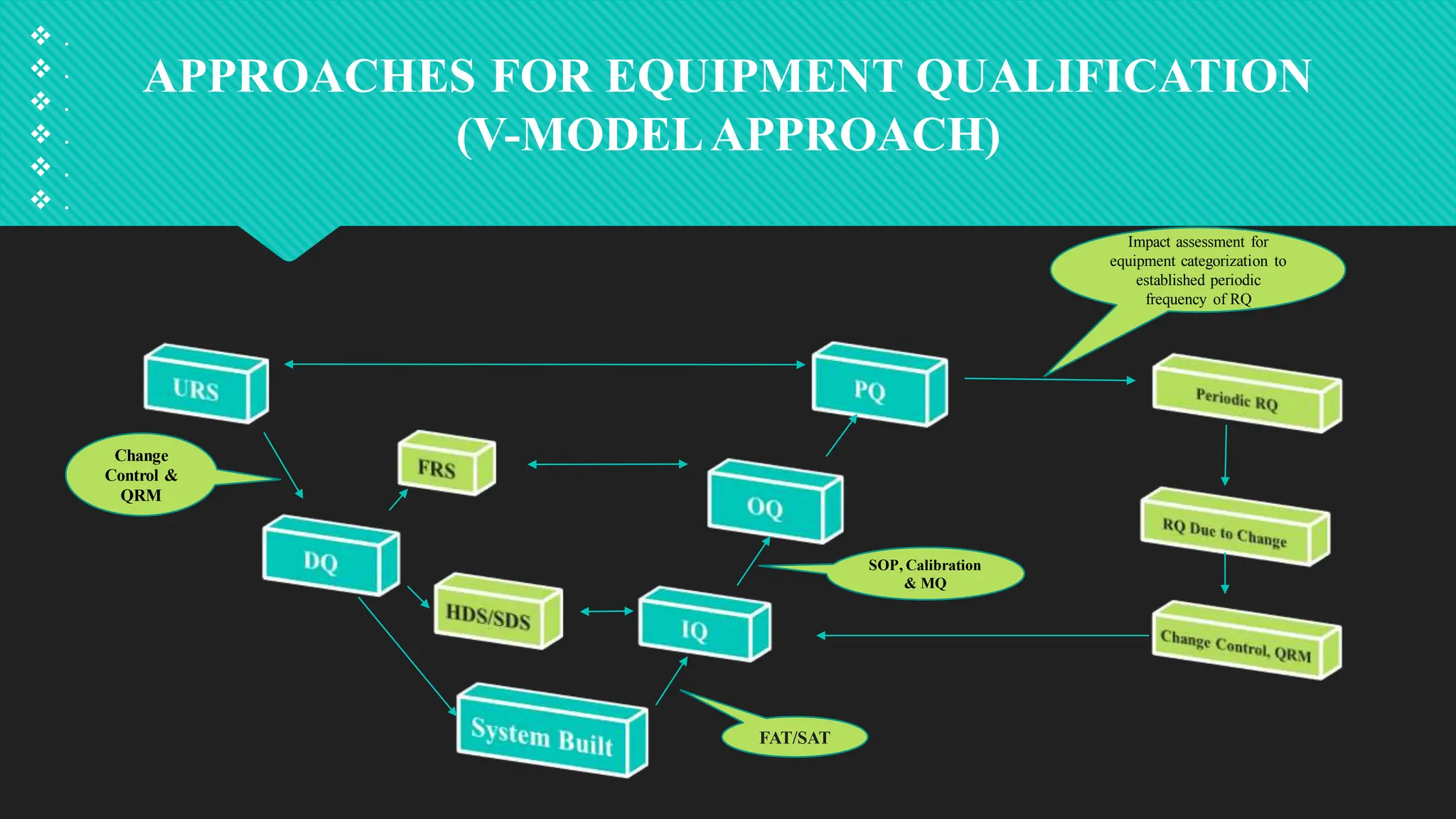
Qualification and validation are processes used to ensure that equipment, systems, processes, and products meet specific standards, requirements, and regulations.
Qualification typically involves:
1. Verifying that equipment, systems, or processes meet specifications and requirements.
2. Documenting evidence of compliance.
Validation involves:
1. Confirming that processes, systems, or products consistently deliver desired outcomes.
2. Ensuring compliance with regulatory requirements.
Together, qualification and validation help organizations:
1. Ensure quality and reliability
2. Mitigate risks
3. Meet regulatory requirements
4. Improve efficiency and effectiveness
These processes are crucial in industries like pharmaceuticals, healthcare, and manufacturing, where quality and safety are paramount.