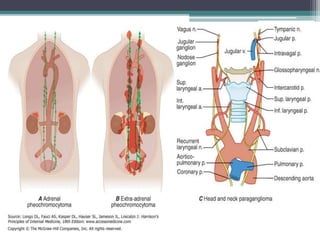
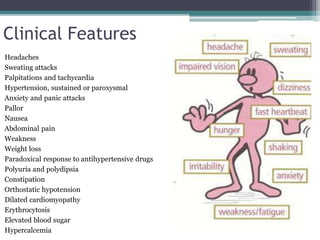
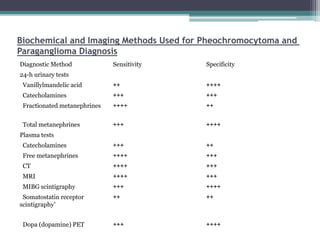
Pheochromocytomas and paragangliomas are catecholamine-producing tumors that can cause hypertension, and their removal can prevent potentially lethal hypertensive crises. Diagnosis is based on the measurement of catecholamines and imaging to locate the tumor, with elevated catecholamine and metanephrine levels indicating a high likelihood of pheochromocytoma. Treatment involves tumor removal and medical management to control blood pressure, including the use of α-adrenergic blockers and possibly β-blockers.
































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)







![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)






