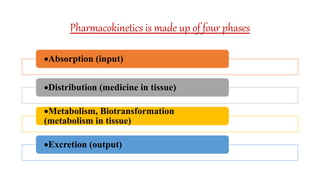
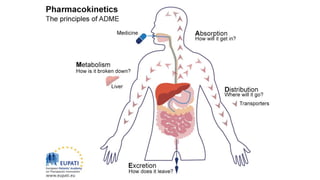
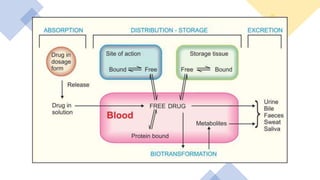
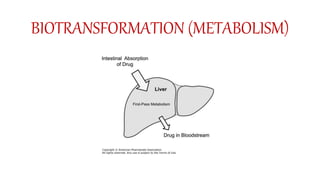
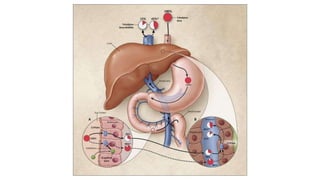
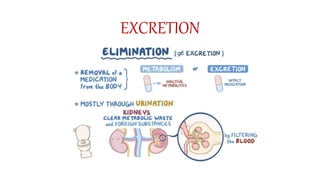
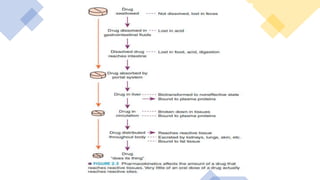
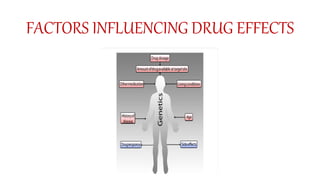
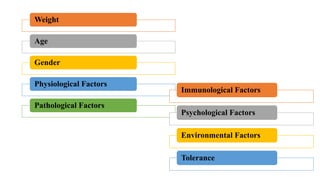
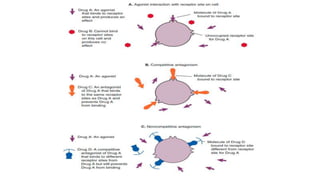
Pharmacokinetics and pharmacodynamics are the two major branches of pharmacology. Pharmacokinetics refers to what the body does to a drug, including absorption, distribution, metabolism, and excretion. Pharmacodynamics refers to what the drug does to the body and its physiological effects. Key concepts in pharmacokinetics include the first-pass effect whereby drugs are metabolized as they pass through the liver, affecting their bioavailability, as well as half-life which determines how long it takes for a drug concentration in the body to reduce by half. Pharmacodynamics examines how drugs act on receptors and enzymes to produce their effects in a selective manner when possible.























![Ideally, all chemotherapeutic agents would act only on enzyme systems that are
essential for the life of a pathogen or neoplastic cell and would not affect healthy
cells. The ability of a drug to attack only those systems found in foreign cells is
known as selective toxicity. Penicillin, an antibiotic used to treat bacterial infections,
has selective toxicity. It affects an enzyme system unique to bacteria, causing
bacterial cell death without disrupting normal human cell functioning.
Unfortunately, most other chemotherapeutic agents also destroy normal human cells,
causing many of the adverse effects associated with antipathogen and antineoplastic
chemotherapy. Cells that reproduce or are replaced rapidly (e.g., bone marrow cells,
gastrointestinal [GI] cells, hair follicles) are more easily affected by these agents.
Consequently, the goal of many chemotherapeutic regimens is to deliver a dose that
will be toxic to the invading cells yet cause the least amount of toxicity to the host.
SELECTIVE TOXICITY](https://image.slidesharecdn.com/pharmacokinetics-220330165714/85/Pharmacokinetics-and-pharmacodynamics-pptx-34-320.jpg)














































































