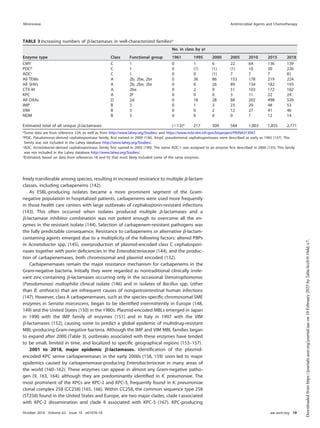
β-lactamases are crucial enzymes responsible for antibiotic resistance in gram-negative bacteria, with origins tracing back millions of years. They have evolved from penicillin-binding proteins and now represent over 2,800 unique proteins, classified into various groups based on their functional and biochemical characteristics. This minireview discusses the historical development of β-lactamases, key classifications, and the ongoing challenges they present in antibiotic therapy.