Download to read offline






































![Difficulty getting relevant description and
datasets from NCBI API using bio* libs
Python example: URL for the Achromyrmex
assembly?
Solution:
ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA_000188075.1_Si_gnG
import xml.etree.ElementTree as ET
from Bio import Entrez
Entrez.email = "mail@bmpvieira.com"
esearch_handle = Entrez.esearch(db="assembly", term="Achromyrmex")
esearch_record = Entrez.read(esearch_handle)
for id in esearch_record['IdList']:
esummary_handle = Entrez.esummary(db="assembly", id=id)
esummary_record = Entrez.read(esummary_handle)
documentSummarySet = esummary_record['DocumentSummarySet']
document = documentSummarySet['DocumentSummary'][0]
metadata_XML = document['Meta'].encode('utf-8')
metadata = ET.fromstring('' + metadata_XML + '')
for entry in Metadata[1]:
print entry.text
bionode-ncbi](https://image.slidesharecdn.com/orcambridge14-150121091315-conversion-gate02/75/Building-collaborative-workflows-for-scientific-data-51-2048.jpg)
![Difficulty getting relevant description and
datasets from NCBI API using bio* libs
Example: URL for the Achromyrmex
assembly?
JavaScript
http://ftp.ncbi.nlm.nih.gov/genomes/all/GCA_000204515.1_Aech_3.9/GCA_00020
4515.1_Aech_3.9_genomic.fna.gz
var bio = require('bionode')
bio.ncbi.urls('assembly', 'Acromyrmex', function(urls) {
console.log(urls[0].genomic.fna)
})
bio.ncbi.urls('assembly', 'Acromyrmex').on('data', printGenomeURL)
function printGenomeURL(urls) {
console.log(urls[0].genomic.fna)
})](https://image.slidesharecdn.com/orcambridge14-150121091315-conversion-gate02/75/Building-collaborative-workflows-for-scientific-data-52-2048.jpg)



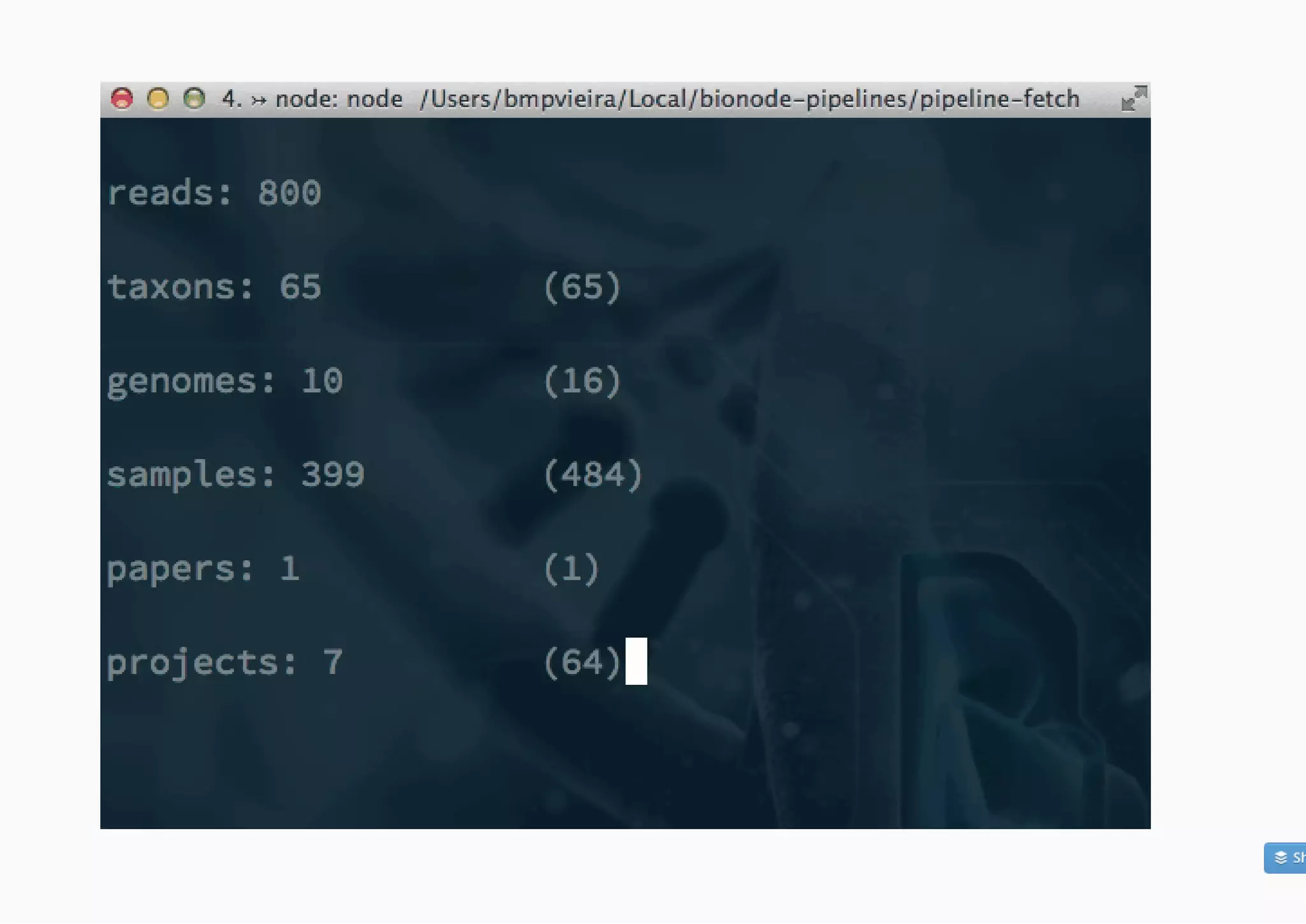


![Difficulty writing scalable, reproducible and
complex bioinformatic pipelines.
{
"import-data": [
"bionode-ncbi search genome eukaryota",
"dat import --json --primary=uid"
],
"search-ncbi": [
"dat cat",
"grep Guillardia",
"tool-stream extractProperty assemblyid",
"bionode-ncbi download assembly -",
"tool-stream collectMatch status completed",
"tool-stream extractProperty uid",
"bionode-ncbi link assembly bioproject -",
"tool-stream extractProperty destUID",
"bionode-ncbi link bioproject sra -",
"tool-stream extractProperty destUID",
"grep 35526",
"bionode-ncbi download sra -",
"tool-stream collectMatch status completed",
"tee > metadata.json"
],](https://image.slidesharecdn.com/orcambridge14-150121091315-conversion-gate02/75/Building-collaborative-workflows-for-scientific-data-60-2048.jpg)
![Difficulty writing scalable, reproducible and
complex bioinformatic pipelines.
"index-and-align": [
"cat metadata.json",
"bionode-sra fastq-dump -",
"tool-stream extractProperty destFile",
"bionode-bwa mem **/*fna.gz"
],
"convert-to-bam": [
"bionode-sam 35526/SRR070675.sam"
]
}](https://image.slidesharecdn.com/orcambridge14-150121091315-conversion-gate02/75/Building-collaborative-workflows-for-scientific-data-61-2048.jpg)








The document discusses the development of collaborative workflows for scientific data management using tools like Dat and Bionode. It addresses challenges in bioinformatics workflows, such as reproducibility and data accessibility, and emphasizes the use of Node.js for building scalable and efficient pipelines. The speaker highlights the importance of open-source tools and community collaboration in advancing scientific research.













































































