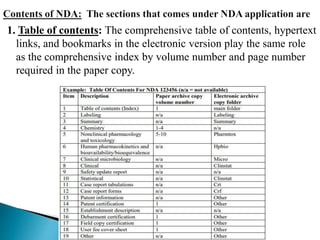
This document provides guidelines for submitting a New Drug Application (NDA) in electronic format to the FDA. It outlines the required sections and contents of an NDA, including: administrative information; drug substance and product information; nonclinical and clinical studies data; safety updates; statistical analysis; and other certifications. The guidelines specify how each section of the application should be organized and formatted, with hyperlinks and bookmarks to aid electronic navigation. All documentation is to be provided in PDF files or datasets within appropriately labeled folders corresponding to each application item number.


























![Drug master file ppt [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/drugmasterfilepptautosaved-200130192621-thumbnail.jpg?width=640&height=640&fit=bounds)

























































