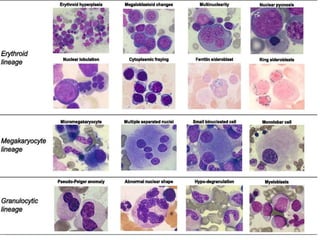
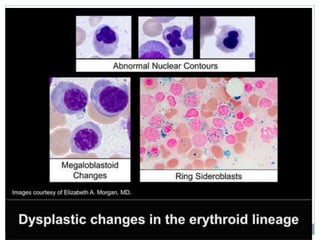
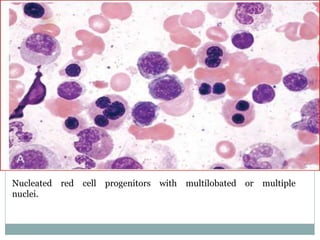
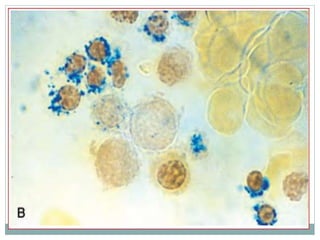
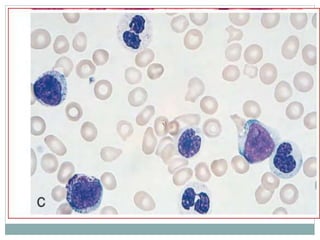
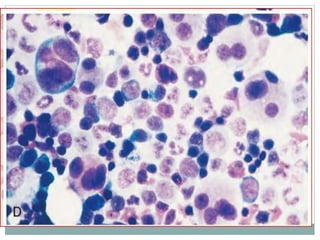
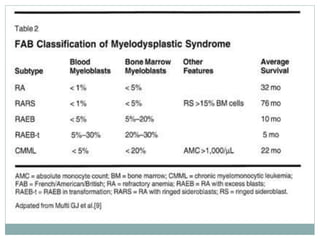
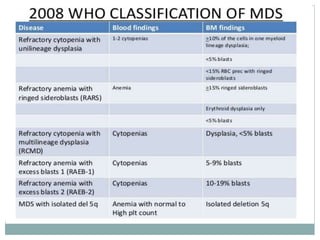
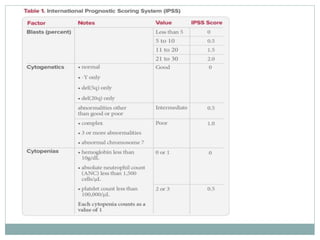
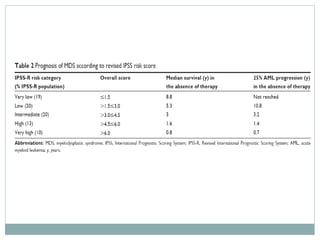
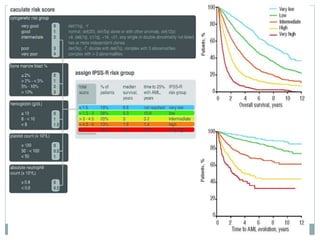
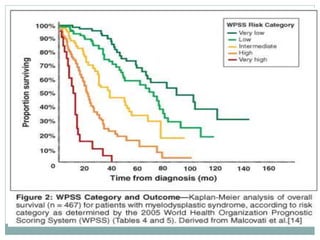
Myelodysplastic syndromes (MDS) are a group of stem cell disorders characterized by ineffective hematopoiesis and a risk of transforming to acute myeloid leukemia. MDS can be primary or secondary to chemotherapy/radiation. The bone marrow shows dysplastic changes in the myeloid lineages. MDS is diagnosed based on blood and bone marrow morphology and cytogenetics. Prognosis depends on blast count, cytopenias, and chromosomal abnormalities, with higher risk features indicating worse outcomes like shorter survival times or faster progression to AML. Treatment options are limited and include stem cell transplantation in younger patients or supportive care in older patients.