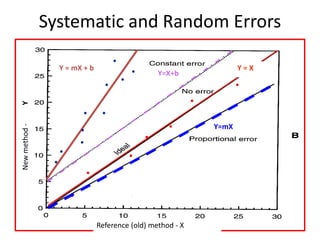
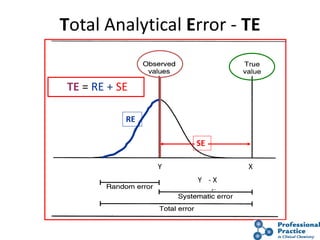
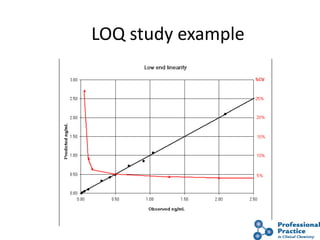
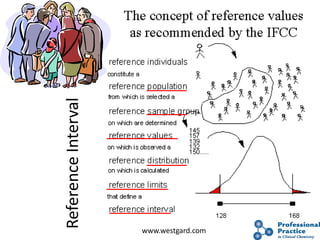
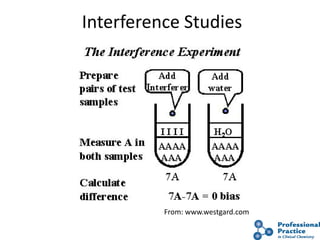
The document outlines the process and steps involved in method validation for clinical testing, emphasizing the importance of defining goals, assessing errors, and comparing performance to analytical standards. It covers key concepts such as total allowable error, analytical measurement range, and the significance of both systematic and random errors. The document serves as a guide for ensuring that new clinical methods meet regulatory requirements and provide accurate medical decisions.