Downloaded 13 times



![• On further probing, she revealed history of recurrent renal stones for the past 16 years. In
addition, she had history of galactorrhea and oligomenorrhea for the past two years. Apart from
this, there was no other significant history. She was the index case in her family.
• However, her family history did reveal that she was part of a large kindred. She had five siblings
and 22 step siblings from the five marriages of her father. Patient’s father who had similar
episodes of neuroglycopenic symptoms [suspected (?) insulinoma] and history of renal stones
had expired before evaluation.
• There was history suggestive of at least one MEN1-associated endocrine tumor among eight of
her siblings.](https://image.slidesharecdn.com/mensyndromesandgeneticbasis-230323102506-cbd76865/75/MEN-SYNDROMESAND-GENETIC-BASIS-pptx-5-2048.jpg)
![• After admission in our ward, the patient had an episode of spontaneous hypoglycemia, and
critical sample was analyzed that revealed non-ketotic hyperinsulinemic hypoglycemia (blood
glucose of 40 mg/dl with corresponding serum insulin 19.4 µU/ml and C peptide of 4.9 ng/ml,
blood ketones of 0.1 mmol/L) [3].
• Serum insulin and C peptide were measured by electrochemiluminescence while blood ketones
were measured on a finger prick test using a point of care device (normal range 0.4-0.5 mmol/L).
• Patient also had parathyroid hormone (PTH)-dependent hypercalcemia with hypercalciuria
[serum total calcium 12.3 mg/dl (normal range: 8.5-10.5 mg/dl), serum intact PTH of 210 pg/ml
(normal range 15-65 pg/ml), 24-hour urinary calcium of 450 mg/day (normal range < 250
mg/day)].
• Her serum prolactin level, measured by ECLIA, was also elevated, 92.6 ng/ml (normal range, 10-
29 ng/ml). Other pituitary hormones were within normal limits.](https://image.slidesharecdn.com/mensyndromesandgeneticbasis-230323102506-cbd76865/75/MEN-SYNDROMESAND-GENETIC-BASIS-pptx-6-2048.jpg)




































![MEN2A with cutaneous lichen
amyloidosis
• CLA (also known as lichen planus amyloidosis [LPA]) has been described in some families with
multiple endocrine neoplasia 2A (MEN2A), predominantly those with the RET codon 634
mutation, although it has also been reported in a patient with a codon 804 mutation .
• The diagnosis of CLA may precede the onset of clinically evident MTC.
• Patients with this variant develop pheochromocytomas and parathyroid hyperplasia with a
similar frequency as those with classical MEN2A.](https://image.slidesharecdn.com/mensyndromesandgeneticbasis-230323102506-cbd76865/75/MEN-SYNDROMESAND-GENETIC-BASIS-pptx-43-2048.jpg)













![Structure and function of the mutated
RET proto-oncogene and its product
• There are differences but much overlap in the specific RET gene mutations underlying the
MEN2A variants and MEN2B.
• Several studies, including one from the International RET Mutation Consortium [25], have
described the relationships between the site of mutation and the type of disease.](https://image.slidesharecdn.com/mensyndromesandgeneticbasis-230323102506-cbd76865/75/MEN-SYNDROMESAND-GENETIC-BASIS-pptx-57-2048.jpg)












![Established disease
• Among patients with multiple endocrine neoplasia type 2 (MEN2), virtually all develop clinically
apparent medullary thyroid cancer (MTC), often early in life .
• Patients with MTC can be cured only by complete resection of the thyroid tumor and any local
and regional metastases.
• ATA (American Thyroid Association) recommend total thyroidectomy for patients with hereditary
forms of MTC (MEN types 2A [MEN2A] and 2B [MEN2B]).](https://image.slidesharecdn.com/mensyndromesandgeneticbasis-230323102506-cbd76865/75/MEN-SYNDROMESAND-GENETIC-BASIS-pptx-70-2048.jpg)



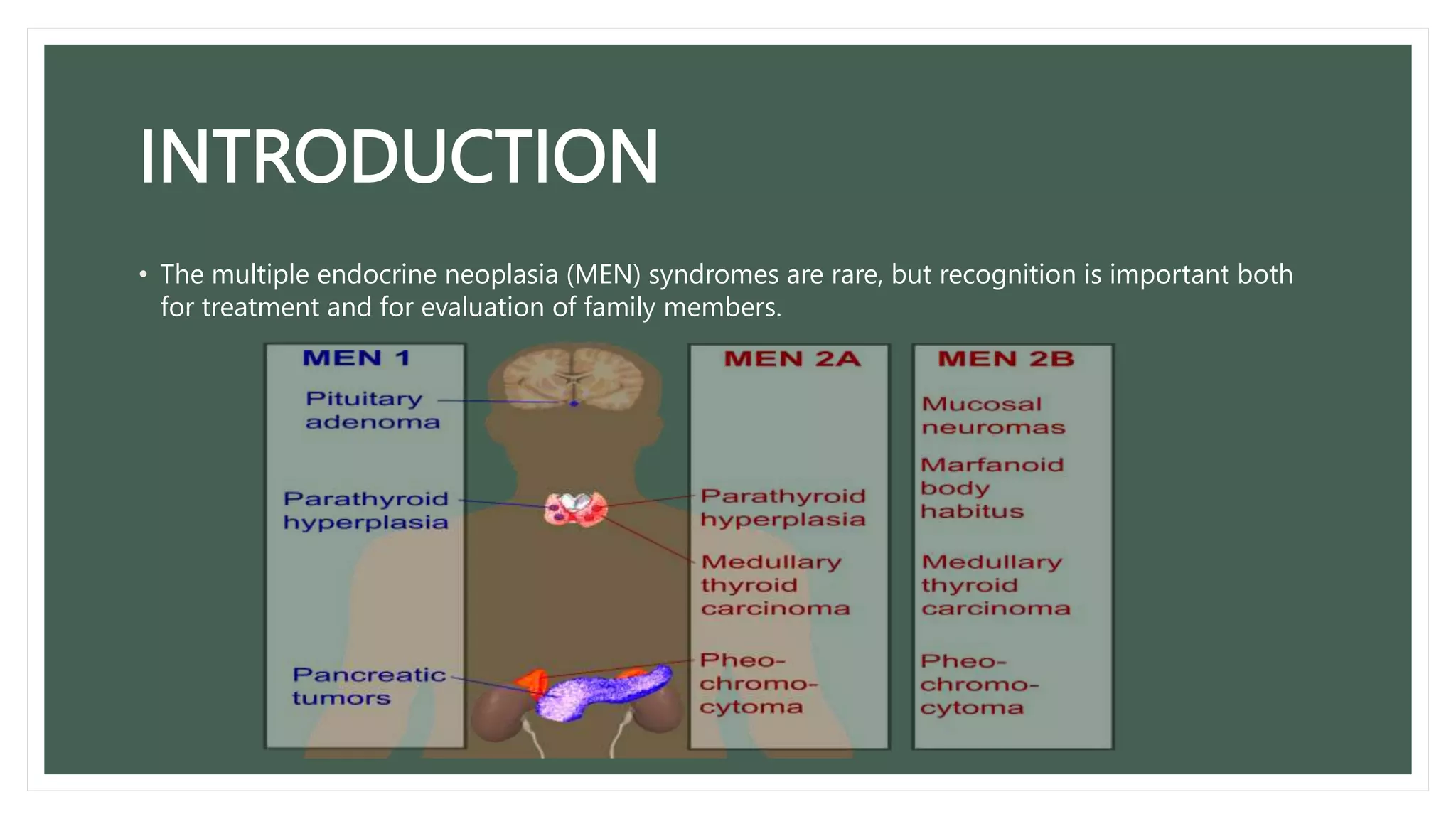
This document discusses a case of multiple endocrine neoplasia type 1 (MEN1) in a 46-year-old female patient and her brother. The patient presented with symptoms of hypoglycemia and was found to have hyperparathyroidism, a pituitary adenoma, and insulinomas. Genetic testing confirmed a MEN1 gene mutation. Her brother also had features of MEN1 including acromegaly, hyperparathyroidism, and insulinomas. MEN1 is a rare genetic disorder characterized by tumors of the parathyroid glands, anterior pituitary, and pancreatic islet cells. Early detection of MEN1-associated tumors through genetic screening and biochemical monitoring of at-risk family members













































![endocrine tumor of pancreas [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/endocrinetumorofpancreasautosaved-241106051359-72ffab89-thumbnail.jpg?width=640&height=640&fit=bounds)






![ivct and ivrt IN CARDIAC CYCLE [Autosaved] .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/ivctandivrtautosaved-copy-250829033609-78e4fe1b-thumbnail.jpg?width=640&height=640&fit=bounds)


































